
Development and Evaluation of a Physiologically Based Pharmacokinetic Model of Labetalol in Healthy and Diseased Populations

 ,
,  ,
,  , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Literature Review
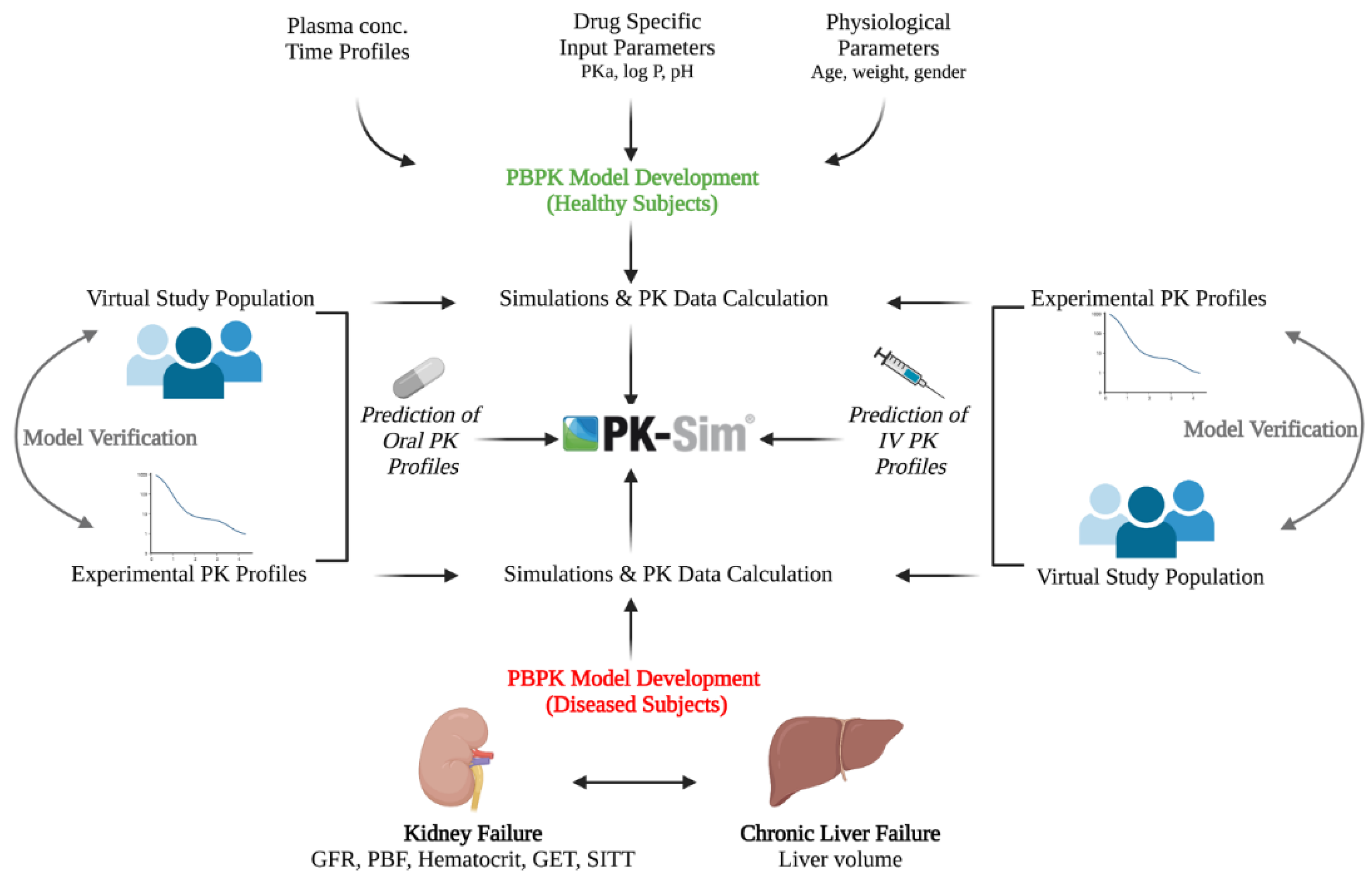
2.2. Modeling Software
2.3. Building Blocks
2.4. Modeling Strategy
2.5. Model Parameters
2.6. Model Structure in Diseased Population
2.6.1. Hepatic Disease
2.6.2. Kidney Disease
2.7. Model Verification
2.8. Influence of Food and Enzymes on Labetalol PK
3. Results
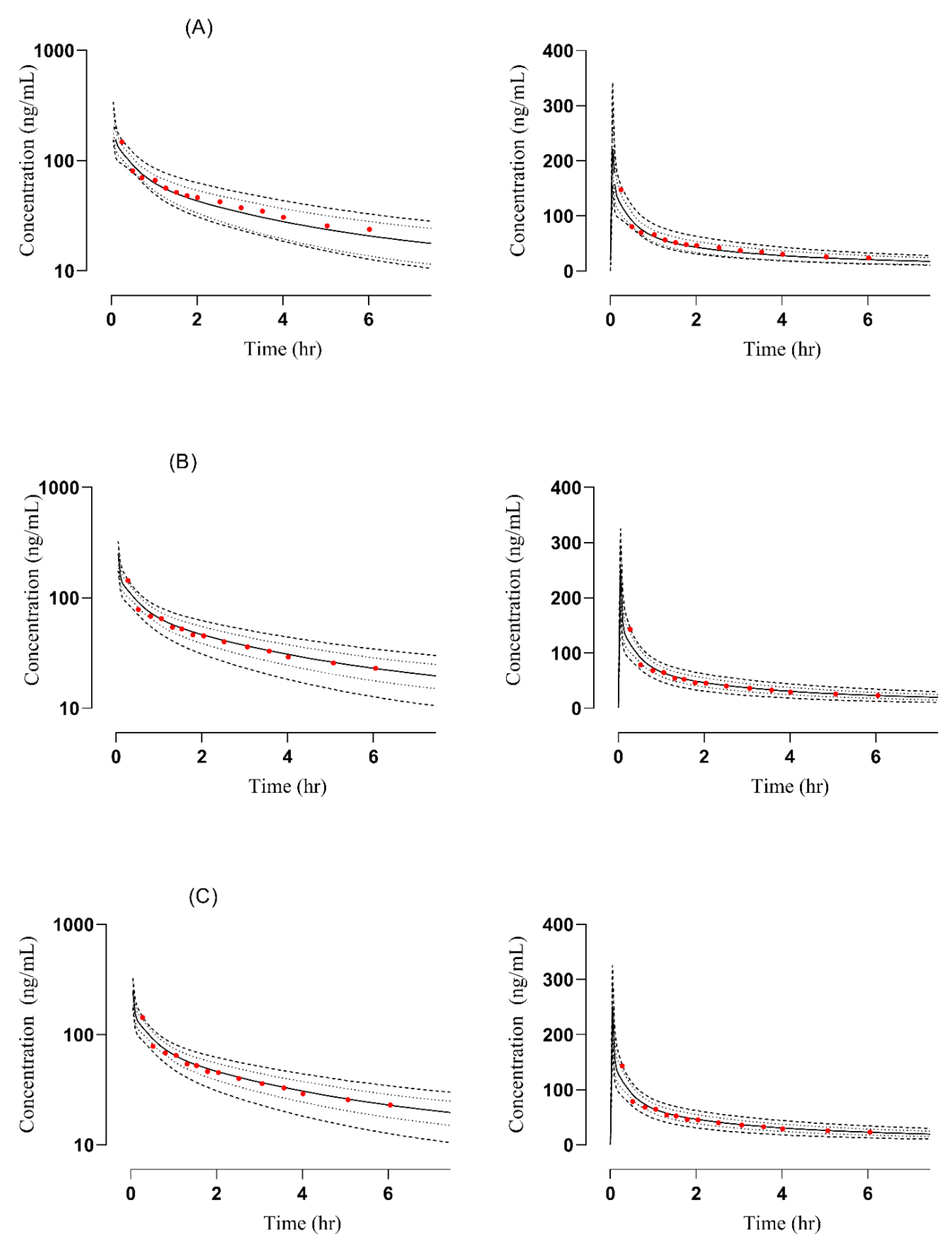
3.1. PBPK Model in Healthy Adults
3.2. PBPK Model in Diseased Population
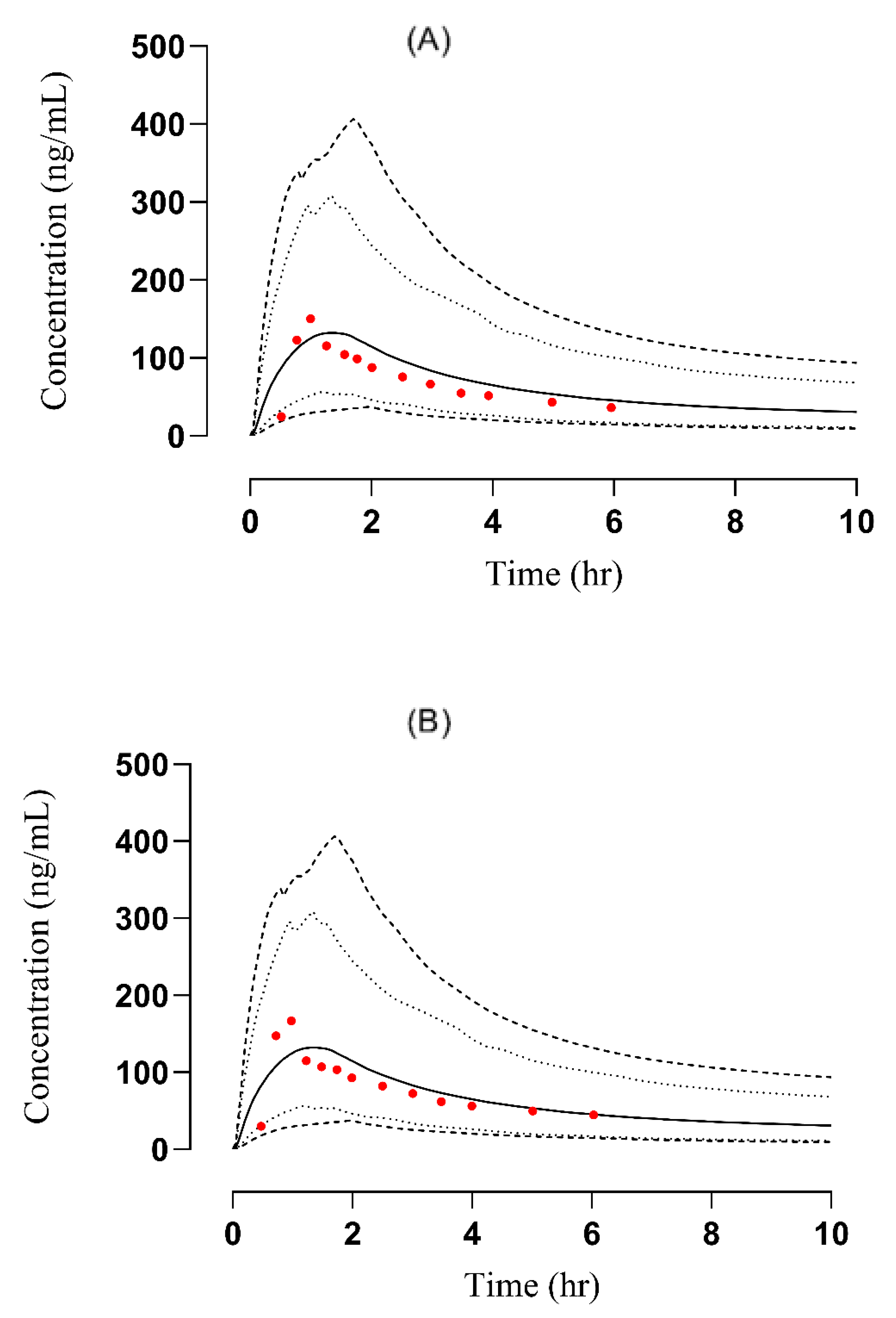
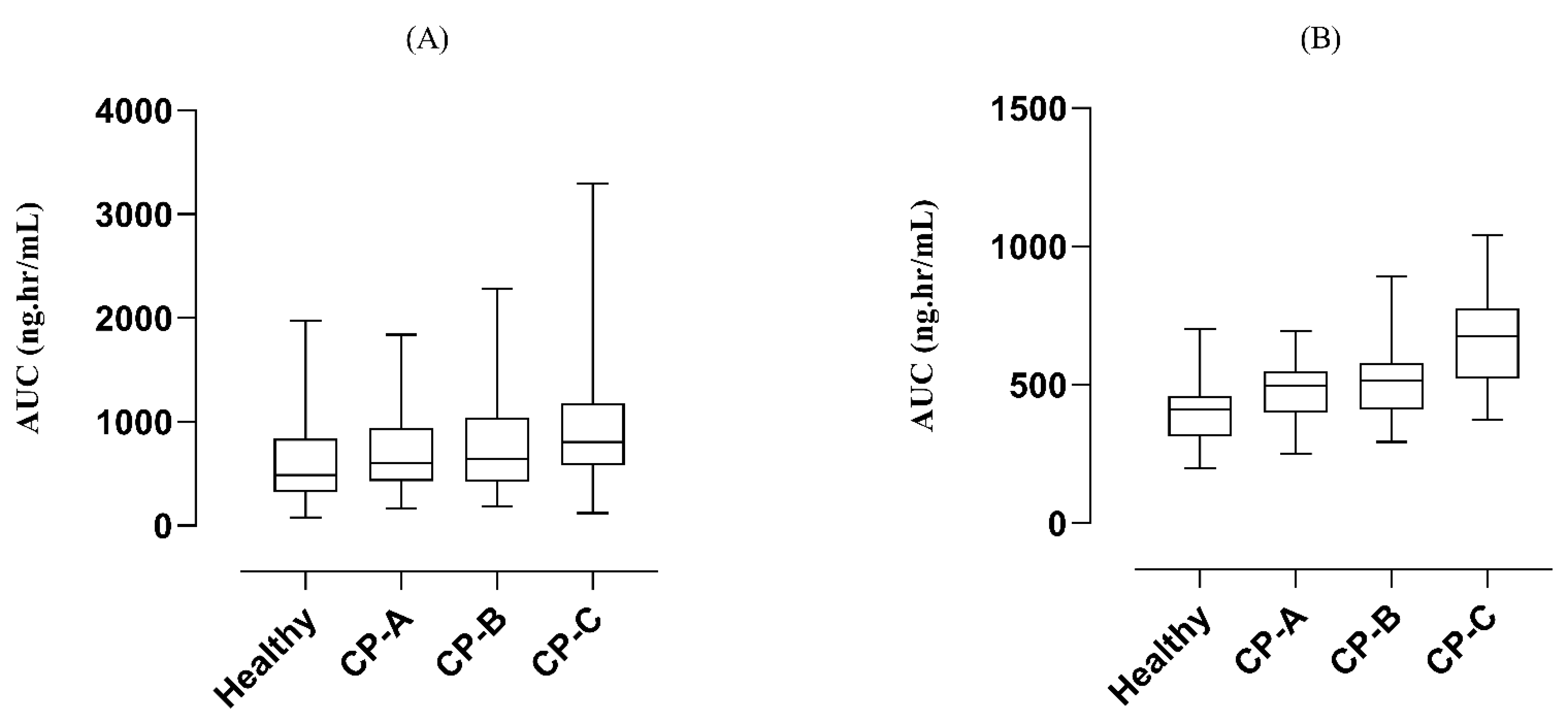
3.2.1. Liver Failure
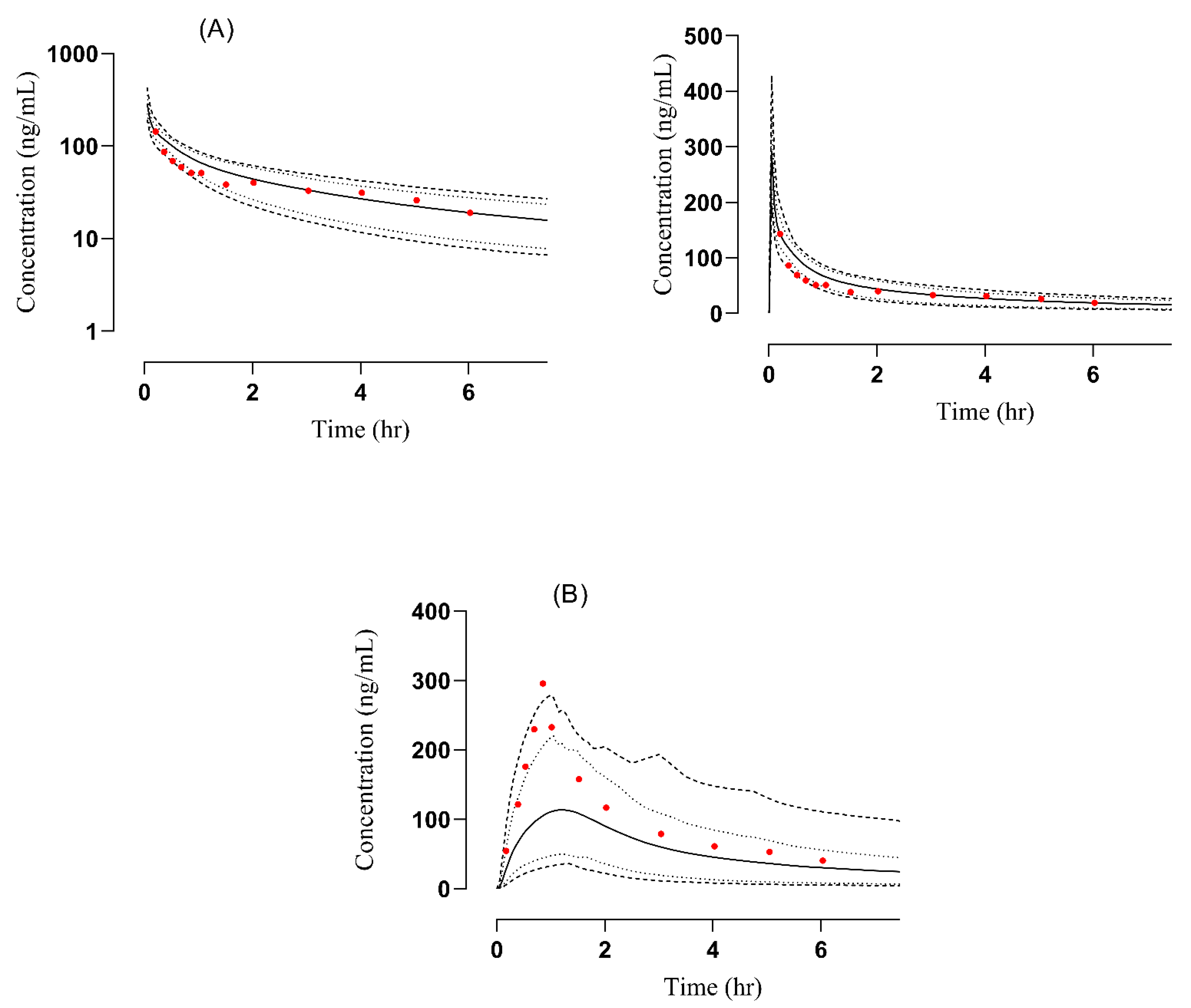
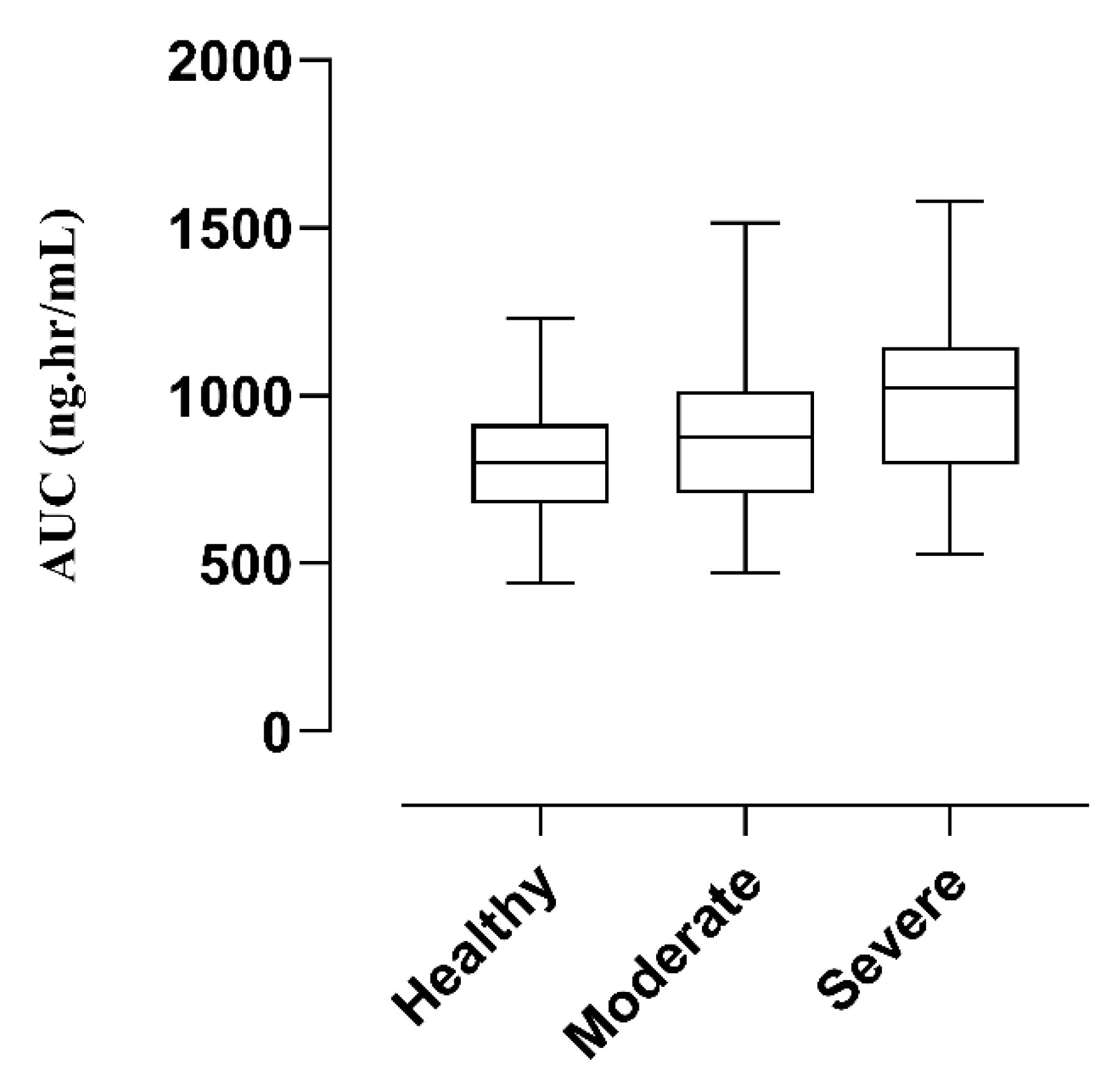
3.2.2. Renal Failure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fischer, J.; Ganelin, C.R. Analogue-based drug discovery. Chem. Int. Newsmag. IUPAC 2010, 32, 12–15. [Google Scholar]
- MacCarthy, E.P.; Bloomfield, S.S. Labetalol: A review of its pharmacology, pharmacokinetics, clinical uses, and adverse effects. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1983, 3, 193–217. [Google Scholar] [CrossRef] [PubMed]
- Lenfant, C.; Chobanian, A.V.; Jones, D.W.; Roccella, E.J. Resetting the hypertension sails. In Proceedings of the Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7). Hypertension 2003, 41, 1178–1179. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, A.; Yusof, M. Labetalol: A Brief Current Review. Pharmacophores 2019, 10, 50–56. [Google Scholar]
- Incecayir, T.; Tsume, Y.; Amidon, G.L. Comparison of the permeability of metoprolol and labetalol in rat, mouse, and Caco-2 cells: Use as a reference standard for BCS classification. Mol. Pharm. 2013, 10, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Peacock, W.F., IV; Hilleman, D.E.; Levy, P.D.; Rhoney, D.H.; Varon, J. A systematic review of nicardipine vs. labetalol for the management of hypertensive crises. Am. J. Emerg. Med. 2012, 30, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Robertson, T.A.; Thorling, C.A.; Zhang, Q.; Fletcher, L.M.; Crawford, D.H.; Roberts, M.S. Hepatic pharmacokinetics of cationic drugs in a high-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Drug Metab. Dispos. 2011, 39, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanto, J.; Allonen, H.; Kleimola, T.; Mäntylä, R. Pharmacokinsetics of labetalol in healthy volunteers. Int. J. Clin. Pharmacol. Ther. Toxicol. 1981, 19, 41–44. [Google Scholar]
- Richards, D.; Maconochie, J.; Bland, R.; Hopkins, R.; Woodings, E.; Martin, L. Relationship between plasma concentrations and pharmacological effects of labetalol. Eur. J. Clin. Pharmacol. 1977, 11, 85–90. [Google Scholar] [CrossRef]
- Doroudian, A. Pharmacokinetics and Conjugative Metabolism of Labetalol Stereoisomers in Pregnant Sheep: A Chiral Drug Case Study in Pregnancy; University of British Columbia: Vancouver, BC, Canada, 1999. [Google Scholar]
- Brogden, R.; Heel, R.; Speight, T.; Avery, G. Labetalol: A review of its pharmacology and therapeutic use in hypertension. Antihypertens. Drugs Today 1979, 4, 100–124. [Google Scholar] [CrossRef]
- Delco, F.; Tchambaz, L.; Schlienger, R.; Drewe, J.; Krähenbühl, S. Dose adjustment in patients with liver disease. Drug Saf. 2005, 28, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Munar, M.Y.; Munar, M.Y.; Signh, H. Drug dosing adjustments in patients with chronic kidney disease. Am. Fam. Physician 2007, 75, 1487–1496. [Google Scholar] [PubMed]
- Cao, Y.; Jusko, W.J. Applications of minimal physiologically-based pharmacokinetic models. J. Pharmacokinet. Pharmacodyn. 2012, 39, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Kuepfer, L.; Niederalt, C.; Wendl, T.; Schlender, J.F.; Willmann, S.; Lippert, J.; Block, M.; Eissing, T.; Teutonico, D. Applied concepts in PBPK modeling: How to build a PBPK/PD model. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 516–531. [Google Scholar] [CrossRef] [Green Version]
- Kostewicz, E.S.; Aarons, L.; Bergstrand, M.; Bolger, M.B.; Galetin, A.; Hatley, O.; Jamei, M.; Lloyd, R.; Pepin, X.; Rostami-Hodjegan, A. PBPK models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 300–321. [Google Scholar] [CrossRef]
- Kalam, M.N.; Rasool, M.F.; Alqahtani, F.; Imran, I.; Rehman, A.U.; Ahmed, N. Development and evaluation of a physiologically based pharmacokinetic drug-disease model of propranolol for suggesting model informed dosing in liver cirrhosis patients. Drug Des. Dev. Ther. 2021, 15, 1195. [Google Scholar] [CrossRef]
- Franchetti, Y.; Nolin, T.D. Dose optimization in kidney disease: Opportunities for PBPK modeling and simulation. J. Clin. Pharmacol. 2020, 60, S36–S51. [Google Scholar] [CrossRef]
- Johnson, T.N.; Boussery, K.; Rowland-Yeo, K.; Tucker, G.T.; Rostami-Hodjegan, A. A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin. Pharmacokinet. 2010, 49, 189–206. [Google Scholar] [CrossRef]
- Rowland Yeo, K.; Aarabi, M.; Jamei, M.; Rostami-Hodjegan, A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev. Clin. Pharmacol. 2011, 4, 261–274. [Google Scholar] [CrossRef]
- Selçuk, A.A. A guide for systematic reviews: Prisma. Turk. Arch. Otorhinolaryngol. 2019, 57, 57. [Google Scholar] [CrossRef]
- Clark, H.D.; Wells, G.A.; Huët, C.; McAlister, F.A.; Salmi, L.R.; Fergusson, D.; Laupacis, A. Assessing the quality of randomized trials: Reliability of the Jadad scale. Control. Clin. Trials 1999, 20, 448–452. [Google Scholar] [CrossRef]
- Al-Dirini, R.M.A.; Thewlis, D.; Paul, G. A comprehensive literature review of the pelvis and the lower extremity FE human models under quasi-static conditions. Work 2012, 41, 4218–4229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneshmend, T.; Roberts, C. The influence of food on the oral and intravenous pharmacokinetics of a high clearance drug: A study with labetalol. Br. J. Clin. Pharmacol. 1982, 14, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Daneshmend, T.; Roberts, C. The effects of enzyme induction and enzyme inhibition on labetalol pharmacokinetics. Br. J. Clin. Pharmacol. 1984, 18, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homeida, M.; Jackson, L.; Roberts, C. Decreased first-pass metabolism of labetalol in chronic liver disease. Br. Med. J. 1978, 2, 1048–1050. [Google Scholar] [CrossRef] [Green Version]
- Wood, A.; Ferry, D.; Bailey, R. Elimination kinetics of labetalol in severe renal failure. Br. J. Clin. Pharmacol. 1982, 13, 81S–86S. [Google Scholar] [CrossRef] [Green Version]
- Willmann, S.; Thelen, K.; Lippert, J. Integration of dissolution into physiologically-based pharmacokinetic models III: PK-Sim®. J. Pharm. Pharmacol. 2012, 64, 997–1007. [Google Scholar] [CrossRef]
- Fischer, J.H.; Sarto, G.E.; Hardman, J.; Endres, L.; Jenkins, T.M.; Kilpatrick, S.J.; Jeong, H.; Geller, S.; Deyo, K.; Fischer, P.A. Influence of gestational age and body weight on the pharmacokinetics of labetalol in pregnancy. Clin. Pharmacokinet. 2014, 53, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Rogers, R.C.; Sibai, B.M.; Whybrew, W.D. Labetalol pharmacokinetics in pregnancy-induced hypertension. Am. J. Obstet. Gynecol. 1990, 162, 362–366. [Google Scholar] [CrossRef]
- Mahmood, I. A Simple Method for the Prediction of Human Concentration–Time Profiles and Pharmacokinetics of Antibody–Drug Conjugates (ADC) from Rats or Monkeys. Antibodies 2022, 11, 42. [Google Scholar] [CrossRef]
- Velenosi, T.J.; Urquhart, B.L. Pharmacokinetic considerations in chronic kidney disease and patients requiring dialysis. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.; Goracci, L.; Cruciani, G.; Poggesi, I. Some considerations on the predictions of pharmacokinetic alterations in subjects with liver disease. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Diep, U.; Chudow, M.; Sunjic, K.M. Pharmacokinetic changes in liver failure and impact on drug therapy. AACN Adv. Crit. Care 2017, 28, 93–101. [Google Scholar]
- Churchwell, M.D.; Mueller, B.A. Selected pharmacokinetic issues in patients with chronic kidney disease. Blood Purif. 2007, 25, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Goa, K.L.; Benfield, P.; Sorkin, E.M. Labetalol. Drugs 1989, 37, 583–627. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. | Population | No. of Subjects | Dose | Portion of Females | Age (years) | Weight (kg) | Reference |
|---|---|---|---|---|---|---|---|
| Oral | |||||||
| 1 | Healthy | 6 | 200 mg | 2 | 20–24 | N/R | [24] |
| 2 | Healthy | 5 | 200 mg | 0 | 21–26 | N/R | [25] |
| IV | |||||||
| 3 | Healthy | 6 | 0.5 mg/kg | 2 | 20–24 | N/R | [24] |
| 4 | Healthy a | 5 | 0.5 mg/kg | 0 | 21–26 | N/R | [25] |
| 5 | Healthy b | 6 | 0.5 mg/kg | 0 | 21–26 | N/R | [25] |
| Diseased | |||||||
| 6 | Chronic liver disease (IV) | 10 | 0.5 mg/kg | 2 | 22–24 | 50–65 | [26] |
| 7 | Chronic liver disease (oral) | 10 | 200 mg | 2 | 22–42 | 50–65 | [26] |
| 8 | Severe renal failure (IV) | 4 | 1 mg/kg | 1 | 37–70 | 75–87 | [27] |
| Parameters | Value | Reference |
|---|---|---|
| Physicochemical parameters | ||
| Molecular weight (g/mol) | 328.412 | PubChem |
| pKa | 9.38 | PubChem |
| Lipophilicity (log units) | 2.79 | PubChem |
| Absorption | ||
| Intestinal permeability (cm/min) | 4.67 × 10−5 | Predicted in PK-Sim |
| Distribution | ||
| Specific organ permeability (L/min) | 0.1 | Optimized value a |
| Fraction unbound (%) | 45 | [29] |
| Partition coefficient model | Pk-Sim standard | |
| Cellular permeability model | Pk-Sim standard | |
| Elimination | ||
| Total body clearance (mL/min/kg) | 15 | [10] |
| Specific clearance (L/h) | 6 | Optimized value a |
| Cmax (ng/mL) a | AUC0–t (ng/mL∙h) b | CL (L/h) c | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IV | ||||||||||
| Study | Observed | Predicted | Ratio | Observed | Predicted | Ratio | Observed | Predicted | Ratio | |
| 1 | 144.244 | 91.9 | 1.56 | 297.002 | 270.74 | 1.09 | 1.2 | 1 | 1.2 | [24] |
| 2 | 143.099 | 120.083 | 1.19 | 299.91 | 288.59 | 1.039 | 0.99 | 1.1 | 0.9 | [25] |
| 3 | 143.09 | 89.178 | 1.60 | 419.66 | 279.972 | 1.498 | 0.7 | 1.1 | 0.63 | [25] |
| Oral | ||||||||||
| 4 | 150.228 | 131.91 | 1.13 | 384.602 | 477.93 | 0.80 | 320 | 270 | 1.18 | [24] |
| 5 | 166.82 | 131.91 | 1.26 | 432.0 | 482.57 | 0.89 | 244 | 260 | 0.93 | [25] |
| Cmax (ng/mL) a | AUC 0–t (ng/mL·h) b | CL (L/h) c | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study | Observed | Predicted | Ratio | Observed | Predicted | Ratio | Observed | Predicted | Ratio | |
| Hepatic IV | ||||||||||
| 1 | 143.18 | 144.1995 | 0.99 | 265.30 | 283.060 | 0.93 | 1.4 | 1.2 | 1.16 | [26] |
| Hepatic Oral | ||||||||||
| 2 | 295.567 | 112.14 | 2.635 | 608.733 | 382.41 | 1.5 | 250.6 | 376 | 0.6 | [26] |
| Renal IV | ||||||||||
| 3 | 535.697 | 588.037 | 0.9109 | 590.26 | 758.806 | 0.77 | 1.4 | 0.98 | 1.5 | [27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafsa, H.; Zamir, A.; Rasool, M.F.; Imran, I.; Saeed, H.; Ahmad, T.; Alsanea, S.; Alshamrani, A.A.; Alruwaili, A.H.; Alqahtani, F. Development and Evaluation of a Physiologically Based Pharmacokinetic Model of Labetalol in Healthy and Diseased Populations. Pharmaceutics 2022, 14, 2362. https://doi.org/10.3390/pharmaceutics14112362
Hafsa H, Zamir A, Rasool MF, Imran I, Saeed H, Ahmad T, Alsanea S, Alshamrani AA, Alruwaili AH, Alqahtani F. Development and Evaluation of a Physiologically Based Pharmacokinetic Model of Labetalol in Healthy and Diseased Populations. Pharmaceutics. 2022; 14(11):2362. https://doi.org/10.3390/pharmaceutics14112362
Chicago/Turabian StyleHafsa, Hafsa, Ammara Zamir, Muhammad Fawad Rasool, Imran Imran, Hamid Saeed, Tanveer Ahmad, Sary Alsanea, Ali A. Alshamrani, Abdullah H. Alruwaili, and Faleh Alqahtani. 2022. "Development and Evaluation of a Physiologically Based Pharmacokinetic Model of Labetalol in Healthy and Diseased Populations" Pharmaceutics 14, no. 11: 2362. https://doi.org/10.3390/pharmaceutics14112362
APA StyleHafsa, H., Zamir, A., Rasool, M. F., Imran, I., Saeed, H., Ahmad, T., Alsanea, S., Alshamrani, A. A., Alruwaili, A. H., & Alqahtani, F. (2022). Development and Evaluation of a Physiologically Based Pharmacokinetic Model of Labetalol in Healthy and Diseased Populations. Pharmaceutics, 14(11), 2362. https://doi.org/10.3390/pharmaceutics14112362