
Innovative Design of Targeted Nanoparticles: Polymer–Drug Conjugates for Enhanced Cancer Therapy



Abstract
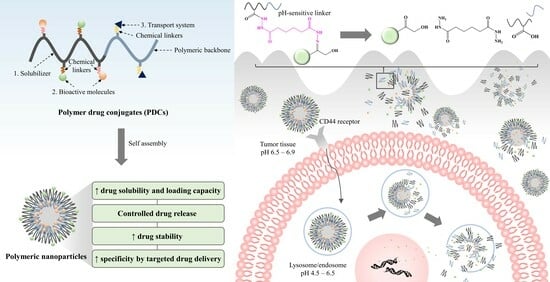
:
1. Introduction
2. Research Progress on PDCs
3. The Principle of PDCs
4. PDC Development for Cancer Treatment
4.1. Modified Physicochemical Properties of Polymers
4.1.1. PCL
4.1.2. TPGS
4.1.3. PEG
4.1.4. HA
4.2. Increased Drug Solubility and Loading Capacity
4.3. Modified Drug Release and Controlled Delivery
4.3.1. pH-Responsive Drug Delivery
4.3.2. Enzyme-Responsive Drug Delivery
4.3.3. Temperature-Responsive Drug Delivery
4.4. Improved Drug Stability under Physiological Conditions
4.5. Increased Specificity through Targeted Drug Delivery of PDCs
4.5.1. Passive Targeting
4.5.2. Active Targeting

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Compositions | Drugs | Ligand | Linkers | Particle Size (nm) | Application | Ref. |
|---|---|---|---|---|---|---|
| Passive targeting | ||||||
| mPEG-PLA | GEM | - | Amide | 112.2 ± 1.86 | Enhanced the efficacy and the stability of blood circulation in the animal model | [10] |
| PEGMA-PLA | CPT | - | Ester | 37.54 | Improved drug stability | [140] |
| PEG | CPT | Ester | 171.9 ± 7.5 | Improved cellular uptake Enhanced cytotoxicity | [139] | |
| MPEG-b-norbornene functional PLA-b-P(α-BrCL) | PTX DOX | - | Ester and amide | 67.8 ± 4.50 | Enhanced the efficacy and synergistic effect | [165] |
| Galactosylated pullulan | CUR | - | Ester | 355 ± 9 | Enhanced cytotoxicity in hepatocellular carcinoma | [166] |
| Acetylated carboxymethylcellulose (Ac-CMC) | Cabazitaxel (CBZ) Docetaxel (DTX) | - | Ester | 96 ± 5.3 | Enhanced cytotoxicity in resistant prostate cancer | [167] |
| Active targeting | ||||||
| PEG-PCL | MTX | Folic acid | Ester | 200–300 | Enhanced cellular uptake | [12] |
| Generation 5 polyamidoamine | MTX | Folic acid | Amide | - | Increased specificity Enhanced cytotoxicity in HeLa cells from cervical carcinoma | [168] |
| HA | DOX GEM | HA | Amide Hydrazone | 20–100 | Increased specificity Enhanced cytotoxicity in a 4T1 orthotopic mouse breast cancer model | [158] |
| HA | GEM CUR | HA | Hydrazone | 221.2 ± 7.7 | Increased specificity Enhanced cytotoxicity in HCT116 and A549 cells | [34] |
| HA | PTX | HA | Ester | - | Enhanced efficacy in mice with bladder cancer | [97] |
| HA | CPT | HA | Amide | - | Improved stability Enhanced cellular uptake | [159] |
| HA | CIS | HA | Ester | - | Enhanced cytotoxicity | [160] |
| PEG | DOX | TTP | - | Increased specificity Enhanced cellular uptake and efficacy | [161] | |
| PLGA-PEG | Trastuzumab (TTP) | TTP | Amide | 81.2 ± 0.9 to 102.5 ± 0.7 | Reduced phagocytic uptake and immunogenicity Increased cellular uptake | [169] |
| PEG-PE | DOX | PEG-pp-PE (MMP-2 sensitive polymer) | Peptide | 33.0 ± 1.2 | Improved multidrug resistance and enhanced efficacy | [163,164] |
| PEG-PLA | Irinotecan (CPT-11) | PEG-pp-PLA (MMP-2 sensitive polymer) | Peptide | 172 ± 30 | Improved multidrug resistance and enhanced efficacy | [162] |
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Cancer. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 10 January 2023).
- Saneja, A.; Kumar, R.; Mintoo, M.J.; Dubey, R.D.; Sangwan, P.L.; Mondhe, D.M.; Panda, A.K.; Gupta, P.N. Gemcitabine and betulinic acid co-encapsulated PLGA−PEG polymer nanoparticles for improved efficacy of cancer chemotherapy. Mater. Sci. Eng. C 2019, 98, 764–771. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Zarrabi, A.; Hashemi, F.; Moghadam, E.R.; Hashemi, F.; Entezari, M.; Hushmandi, K.; Mohammadinejad, R.; Najafi, M. Curcumin in cancer therapy: A novel adjunct for combination chemotherapy with paclitaxel and alleviation of its adverse effects. Life Sci. 2020, 256, 117984. [Google Scholar] [CrossRef] [PubMed]
- Jatzkewitz, H. An ein kolloidales Blutplasma-Ersatzmittel (Polyvinylpyrrolidon) gebundenes Peptamin (Glycyl-L-leucyl-mezcalin) als neuartige Depotform für biologisch aktive primäre Amine (Mezcalin). Z. Für Nat. B 1955, 10, 27. [Google Scholar] [CrossRef]
- Thakor, P.; Bhavana, V.; Sharma, R.; Srivastava, S.; Singh, S.B.; Mehra, N.K. Polymer–drug conjugates: Recent advances and future perspectives. Drug Discov. Today 2020, 25, 1718–1726. [Google Scholar] [CrossRef]
- Basu, S.; Das, A. Curcumin Hyaluronan-Compounds. Available online: http://www.google.com/patents/US20090170195 (accessed on 15 July 2023).
- Manju, S.; Sreenivasan, K. Conjugation of curcumin onto hyaluronic acid enhances its aqueous solubility and stability. J. Colloid Interface Sci. 2011, 359, 318–325. [Google Scholar] [CrossRef]
- Huang, X.; Liao, W.; Xie, Z.; Chen, D.; Zhang, C.Y. A pH-responsive prodrug delivery system self-assembled from acid-labile doxorubicin-conjugated amphiphilic pH-sensitive block copolymers. Mater. Sci. Eng. C 2018, 90, 27–37. [Google Scholar] [CrossRef]
- Xiong, D.; Zhang, X.; Peng, S.; Gu, H.; Zhang, L. Smart pH-sensitive micelles based on redox degradable polymers as DOX/GNPs carriers for controlled drug release and CT imaging. Colloids Surf. B Biointerfaces 2018, 163, 29–40. [Google Scholar] [CrossRef]
- Liang, T.-J.; Zhou, Z.-M.; Cao, Y.-Q.; Ma, M.-Z.; Wang, X.-J.; Jing, K. Gemcitabine-based polymer-drug conjugate for enhanced anticancer effect in colon cancer. Int. J. Pharm. 2016, 513, 564–571. [Google Scholar] [CrossRef]
- Huang, D.; Zhuang, Y.; Shen, H.; Yang, F.; Wang, X.; Wu, D. Acetal-linked PEGylated paclitaxel prodrugs forming free-paclitaxel-loaded pH-responsive micelles with high drug loading capacity and improved drug delivery. Mater. Sci. Eng. C 2018, 82, 60–68. [Google Scholar] [CrossRef]
- Issarachot, O.; Suksiriworapong, J.; Takano, M.; Yumoto, R.; Junyaprasert, V.B. Folic acid-modified methotrexate-conjugated PEGylated poly(ε-caprolactone) nanoparticles for targeted delivery. J. Nanoparticle Res. 2014, 16, 2276. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, F.; Song, X.; Wu, J.; Yao, W.; Gao, X. Conjugation of paclitaxel to C-6 hexanediamine-modified hyaluronic acid for targeted drug delivery to enhance antitumor efficacy. Carbohydr. Polym. 2018, 181, 150–158. [Google Scholar] [CrossRef]
- Suksiriworapong, J.; Sripha, K.; Kreuter, J.; Junyaprasert, V.B. Functionalized (poly(↋-caprolactone))2-poly(ethylene glycol) nanoparticles with grafting nicotinic acid as drug carriers. Int. J. Pharm. 2012, 423, 562–570. [Google Scholar] [CrossRef]
- Anitha, A.; Deepa, N.; Chennazhi, K.P.; Lakshmanan, V.-K.; Jayakumar, R. Combinatorial anticancer effects of curcumin and 5-fluorouracil loaded thiolated chitosan nanoparticles towards colon cancer treatment. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 2730–2743. [Google Scholar] [CrossRef]
- Mathe, G.; Loc, T.B.; Bernard, J. Effet sur la leucemie LI210 de la souris d’une combinaison par diazotation d’A-methopterine et de gamma-globulines de hamsters porteurs de cette leucemie par heterogreffe. Comtes Rendus Hebd. Seances Acad. Sci. 1958, 246, 1626–1628. [Google Scholar]
- Givental, N.I.; Ushakov, S.N.; Panarin, E.F.; Popova, G.O. Experimental studies on penicillin polymer derivatives. Antibiotiki 1965, 10, 701–706. [Google Scholar] [PubMed]
- De Duve, C.; De Barsy, T.; Poole, B.; Trouet, A.; Tulkens, P.; Van Hoof, F.o. Lysosomotropic agents. Biochem. Pharmacol. 1974, 23, 2495–2531. [Google Scholar] [CrossRef]
- Ringsdorf, H. Structure and properties of pharmacologically active polymers. J. Polym. Sci. Polym. Symp. 1975, 51, 135–153. [Google Scholar]
- Abuchowski, A.; McCoy, J.R.; Palczuk, N.C.; van Es, T.; Davis, F.F. Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J. Biol. Chem. 1977, 252, 3582–3586. [Google Scholar]
- Abuchowski, A.; van Es, T.; Palczuk, N.C.; Davis, F.F. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J. Biol. Chem. 1977, 252, 3578–3581. [Google Scholar]
- Vasey, P.A.; Kaye, S.B.; Morrison, R.; Twelves, C.; Wilson, P.; Duncan, R.; Thomson, A.H.; Murray, L.S.; Hilditch, T.E.; Murray, T.; et al. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: First member of a new class of chemotherapeutic agents-drug-polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clin. Cancer Res. 1999, 5, 83–94. [Google Scholar]
- Malugin, A.; Kopečková, P.; Kopeček, J. Liberation of doxorubicin from HPMA copolymer conjugate is essential for the Induction of cell cycle arrest and nuclear fragmentation in ovarian carcinoma cells. J. Control. Release Off. J. Control. Release Soc. 2007, 124, 6–10. [Google Scholar] [CrossRef]
- Seymour, L.W.; Ferry, D.R.; Kerr, D.J.; Rea, D.; Whitlock, M.; Poyner, R.; Boivin, C.; Hesslewood, S.; Twelves, C.; Blackie, R.; et al. Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer. Int. J. Oncol. 2009, 34, 1629–1636. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Feng, Q.; Tong, R. Anticancer nanoparticulate polymer-drug conjugate. Bioeng. Transl. Med. 2016, 1, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Javia, A.; Vanza, J.; Bardoliwala, D.; Ghosh, S.; Misra, L.A.; Patel, M.; Thakkar, H. Polymer-drug conjugates: Design principles, emerging synthetic strategies and clinical overview. Int. J. Pharm. 2022, 623, 121863. [Google Scholar] [CrossRef]
- Yang, J.; Kopeček, J. Design of smart HPMA copolymer-based nanomedicines. J. Control. Release 2016, 240, 9–23. [Google Scholar] [CrossRef]
- Maeda, H.; Takeshita, J.; Kanamaru, R. A lipophilic derivative of neocarzinostatin. A polymer conjugation of an antitumor protein antibiotic. Int. J. Pept. Protein Res. 1979, 14, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, A.R. Pegaspargase (oncaspar). J. Pediatr. Oncol. Nurs. 1995, 12, 46–48. [Google Scholar] [CrossRef]
- Segal, E.; Pan, H.; Benayoun, L.; Kopečková, P.; Shaked, Y.; Kopeček, J.; Satchi-Fainaro, R. Enhanced anti-tumor activity and safety profile of targeted nano-scaled HPMA copolymer-alendronate-TNP-470 conjugate in the treatment of bone malignances. Biomaterials 2011, 32, 4450–4463. [Google Scholar] [CrossRef]
- Fu, S.; Rempson, C.M.; Puche, V.; Zhao, B.; Zhang, F. Construction of disulfide containing redox-responsive polymeric nanomedicine. Methods 2022, 199, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Seifu, M.F.; Nath, L.K. Polymer-drug conjugates: Novel carriers for cancer chemotherapy. Polym.-Plast. Technol. Mater. 2019, 58, 158–171. [Google Scholar] [CrossRef]
- Thummarati, P.; Suksiriworapong, J.; Sakchaisri, K.; Nawroth, T.; Langguth, P.; Roongsawang, B.; Junyaprasert, V.B. Comparative study of dual delivery of gemcitabine and curcumin using CD44 targeting hyaluronic acid nanoparticles for cancer therapy. J. Drug Deliv. Sci. Technol. 2022, 77, 103883. [Google Scholar] [CrossRef]
- Deirram, N.; Zhang, C.; Kermaniyan, S.S.; Johnston, A.P.R.; Such, G.K. pH-responsive polymer nanoparticles for drug delivery. Macromol. Rapid Commun. 2019, 40, 1800917. [Google Scholar] [CrossRef]
- Wang, M.; Yan, J.; Li, C.; Wang, X.; Xiong, J.; Pan, D.; Wang, L.; Xu, Y.; Li, X.; Yang, M. Cationic poly(amide-imide)-conjugated camptothecin prodrug with variable nanomorphology for efficient reductive-responsive drug delivery. Eur. Polym. J. 2020, 123, 109462. [Google Scholar] [CrossRef]
- Li, M.; Gao, M.; Fu, Y.; Chen, C.; Meng, X.; Fan, A.; Kong, D.; Wang, Z.; Zhao, Y. Acetal-linked polymeric prodrug micelles for enhanced curcumin delivery. Colloids Surf. B Biointerfaces 2016, 140, 11–18. [Google Scholar] [CrossRef]
- Van Natta, F.J.; Hill, J.W.; Carothers, W.H. Studies of polymerization and ring formation. XXIII. ↋-caprolactone and its polymers. J. Am. Chem. Soc. 1934, 56, 455–457. [Google Scholar] [CrossRef]
- Kaluzynski, K.; Pretula, J.; Lewinski, P.; Kaźmierski, S.; Penczek, S. Synthesis and properties of functionalized poly(ε-caprolactone); chain polymerization followed by polycondensation in one pot with initiator and catalyst in one molecule. synthesis and molecular structures. Macromolecules 2022, 55, 2210–2221. [Google Scholar] [CrossRef]
- Rai, A.; Senapati, S.; Saraf, S.K.; Maiti, P. Biodegradable poly(ε-caprolactone) as a controlled drug delivery vehicle of vancomycin for the treatment of MRSA infection. J. Mater. Chem. B 2016, 4, 5151–5160. [Google Scholar] [CrossRef]
- Huang, M.-H.; Li, S.; Coudane, J.; Vert, M. Synthesis and characterization of block copolymers of ε-caprolactone and DL-lactide initiated by ethylene glycol or poly(ethylene glycol). Macromol. Chem. Phys. 2003, 204, 1994–2001. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef]
- Manivasagam, G.; Reddy, A.; Sen, D.; Nayak, S.; Mathew, M.T.; Rajamanikam, A. Dentistry: Restorative and Regenerative Approaches. In Encyclopedia of Biomedical Engineering; Narayan, R., Ed.; Elsevier: Oxford, UK, 2019; pp. 332–347. [Google Scholar]
- Suksiriworapong, J.; Phoca, K.; Ngamsom, S.; Sripha, K.; Moongkarndi, P.; Junyaprasert, V.B. Comparison of poly(ε-caprolactone) chain lengths of poly(ε-caprolactone)-co-d-α-tocopheryl-poly(ethylene glycol) 1000 succinate nanoparticles for enhancement of quercetin delivery to SKBR3 breast cancer cells. Eur. J. Pharm. Biopharm. 2016, 101, 15–24. [Google Scholar] [CrossRef]
- Ponjavic, M.; Nikolic, M.S.; Nikodinovic-Runic, J.; Jeremic, S.; Stevanovic, S.; Djonlagic, J. Degradation behaviour of PCL/PEO/PCL and PCL/PEO block copolymers under controlled hydrolytic, enzymatic and composting conditions. Polym. Test. 2017, 57, 67–77. [Google Scholar] [CrossRef]
- Pisani, S.; Dorati, R.; Conti, B.; Modena, T.; Bruni, G.; Genta, I. Design of copolymer PLA-PCL electrospun matrix for biomedical applications. React. Funct. Polym. 2018, 124, 77–89. [Google Scholar] [CrossRef]
- Guo, Z.-w.; Gallo, J.M. Selective Protection of 2‘,2‘-Difluorodeoxycytidine (Gemcitabine). J. Org. Chem. 1999, 64, 8319–8322. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tan, B.H. Towards the development of polycaprolactone based amphiphilic block copolymers: Molecular design, self-assembly and biomedical applications. Mater. Sci. Eng. C 2014, 45, 620–634. [Google Scholar] [CrossRef]
- Al Samad, A.; Bethry, A.; Koziolova, E.; Netopilik, M.; Etrych, T.; Bakkour, Y.; Coudane, J.; El Omar, F.; Nottelet, B. PCL-PEG graft copolymers with tunable amphiphilicity as efficient drug delivery systems. J. Mater. Chem. B 2016, 4, 6228–6239. [Google Scholar] [CrossRef]
- Issarachot, O.; Suksiriworapong, J.; Sripha, K.; Junyaprasert, V. Modification of tricomponent and dicomponent poly(ε-caprolactone)-co-poly(ethylene glycol) with methotrexate and folic acid. J. Appl. Polym. Sci. 2013, 129, 721–734. [Google Scholar] [CrossRef]
- Pawar, R.; Pathan, A.; Nagaraj, S.; Kapare, H.; Giram, P.; Wavhale, R. Polycaprolactone and its derivatives for drug delivery. Polym. Adv. Technol. n/a. [CrossRef]
- Li, H.Y.; Zhang, B.; Chan, P.S.; Weng, J.; Tsang, C.K.; Lee, W.Y.T. Convergent synthesis and characterization of fatty acid-conjugated poly(ethylene glycol)-block-poly(epsilon-caprolactone) nanoparticles for improved drug delivery to the brain. Eur. Polym. J. 2018, 98, 394–401. [Google Scholar] [CrossRef]
- Hailemeskel, B.Z.; Hsu, W.-H.; Addisu, K.D.; Andrgie, A.T.; Chou, H.-Y.; Lai, J.-Y.; Tsai, H.-C. Diselenide linkage containing triblock copolymer nanoparticles based on Bi(methoxyl poly(ethylene glycol))-poly(ε-carprolactone): Selective intracellular drug delivery in cancer cells. Mater. Sci. Eng. C 2019, 103, 109803. [Google Scholar] [CrossRef]
- Guo, Y.; Luo, J.; Tan, S.; Otieno, B.O.; Zhang, Z. The applications of Vitamin E TPGS in drug delivery. Eur. J. Pharm. Sci. 2013, 49, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Beig, A.; Fine-Shamir, N.; Porat, D.; Lindley, D.; Miller, J.M.; Dahan, A. Concomitant solubility-permeability increase: Vitamin E TPGS vs. amorphous solid dispersion as oral delivery systems for etoposide. Eur. J. Pharm. Biopharm. 2017, 121, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Ishak, R.A.H.; Osman, R. Lecithin/TPGS-based spray-dried self-microemulsifying drug delivery systems: In vitro pulmonary deposition and cytotoxicity. Int. J. Pharm. 2015, 485, 249–260. [Google Scholar] [CrossRef]
- Shao, Y.; Yang, L.; Han, H.-K. TPGS-chitosome as an effective oral delivery system for improving the bioavailability of Coenzyme Q10. Eur. J. Pharm. Biopharm. 2015, 89, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, B.; Wang, G.; Wu, Y.; Zhang, X.; Chen, Y.; Tang, S.; Yuan, Y.; Lee, R.J.; Teng, L.; et al. Enhanced antitumor efficacy of vitamin E TPGS-emulsified PLGA nanoparticles for delivery of paclitaxel. Colloids Surf. B Biointerfaces 2014, 123, 716–723. [Google Scholar] [CrossRef]
- Beilman, J.; Blakeley, R.V.B.; Strong, D.B. Tissue and excrement distribution kinetics following a single oral dose of tocopheryl (14C) polyethylene glycol 1000 in rats. Eastman Pharm. Tech. Rep. 2007, 490, 1–20. [Google Scholar]
- Mi, Y.; Zhao, J.; Feng, S.S. Vitamin E TPGS prodrug micelles for hydrophilic drug delivery with neuroprotective effects. Int. J. Pharm. 2012, 438, 98–106. [Google Scholar] [CrossRef]
- Junyaprasert, V.B.; Dhanahiranpruk, S.; Suksiriworapong, J.; Sripha, K.; Moongkarndi, P. Enhanced toxicity and cellular uptake of methotrexate-conjugated nanoparticles in folate receptor-positive cancer cells by decorating with folic acid-conjugated d-α-tocopheryl polyethylene glycol 1000 succinate. Colloids Surf. B Biointerfaces 2015, 136, 383–393. [Google Scholar] [CrossRef]
- Alshetaili, A.S.; Ali, R.; Qamar, W.; Almohizea, S.; Anwer, M.K. Preparation, optimization, and characterization of chrysin-loaded TPGS-b-PCL micelles and assessment of their cytotoxic potential in human liver cancer (Hep G2) cell lines. Int. J. Biol. Macromol. 2023, 246, 125679. [Google Scholar] [CrossRef]
- Agrawal, P.; Sonali; Singh, R.P.; Sharma, G.; Mehata, A.K.; Singh, S.; Rajesh, C.V.; Pandey, B.L.; Koch, B.; Muthu, M.S. Bioadhesive micelles of d-alpha-tocopherol polyethylene glycol succinate 1000: Synergism of chitosan and transferrin in targeted drug delivery. Colloids Surfaces. B Biointerfaces 2017, 152, 277–288. [Google Scholar] [CrossRef]
- Mehata, A.K.; Setia, A.; Vikas; Malik, A.K.; Hassani, R.; Dailah, H.G.; Alhazmi, H.A.; Albarraq, A.A.; Mohan, S.; Muthu, M.S. Vitamin E TPGS-based nanomedicine, nanotheranostics, and targeted drug delivery: Past, present, and future. Pharmaceutics 2023, 15, 722. [Google Scholar] [CrossRef] [PubMed]
- Collnot, E.M.; Baldes, C.; Wempe, M.F.; Kappl, R.; Huttermann, J.; Hyatt, J.A.; Edgar, K.J.; Schaefer, U.F.; Lehr, C.M. Mechanism of inhibition of P-glycoprotein mediated efflux by vitamin E TPGS: Influence on ATPase activity and membrane fluidity. Mol. Pharm. 2007, 4, 465–474. [Google Scholar] [CrossRef]
- Youk, H.-J.; Lee, E.; Choi, M.-K.; Lee, Y.-J.; Chung, J.H.; Kim, S.-H.; Lee, C.-H.; Lim, S.-J. Enhanced anticancer efficacy of α-tocopheryl succinate by conjugation with polyethylene glycol. J. Control. Release 2005, 107, 43–52. [Google Scholar] [CrossRef]
- Almawash, S.; Chaturvedi, S.; Misra, C.; Thotakura, N.; Ibrahim, I.M.; Sharma, G.; Katare, O.P.; Preet, S.; Raza, K. Vitamin E TPGS-PLGA-based nanoparticles for methotrexate delivery: Promising outcomes from preclinical studies. J. Drug Deliv. Sci. Technol. 2022, 72, 103276. [Google Scholar] [CrossRef]
- Gan, H.; Chen, L.; Sui, X.; Wu, B.; Zou, S.; Li, A.; Zhang, Y.; Liu, X.; Wang, D.; Cai, S.; et al. Enhanced delivery of sorafenib with anti-GPC3 antibody-conjugated TPGS-b-PCL/Pluronic P123 polymeric nanoparticles for targeted therapy of hepatocellular carcinoma. Mater. Sci. Eng. C 2018, 91, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-R.; Yang, S.-Y.; Chen, G.-X.; Wei, P. Barbaloin loaded polydopamine-polylactide-TPGS (PLA-TPGS) nanoparticles against gastric cancer as a targeted drug delivery system: Studies in vitro and in vivo. Biochem. Biophys. Res. Commun. 2018, 499, 8–16. [Google Scholar] [CrossRef]
- Cao, N.; Feng, S.S. Doxorubicin conjugated to D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS): Conjugation chemistry, characterization, in vitro and in vivo evaluation. Biomaterials 2008, 29, 3856–3865. [Google Scholar] [CrossRef]
- Khare, V.; Sakarchi, W.A.; Gupta, P.N.; Curtis, A.D.M.; Hoskins, C. Synthesis and characterization of TPGS–gemcitabine prodrug micelles for pancreatic cancer therapy. RSC Adv. 2016, 6, 60126–60137. [Google Scholar] [CrossRef]
- Kutty, R.V.; Feng, S.-S. Cetuximab conjugated vitamin E TPGS micelles for targeted delivery of docetaxel for treatment of triple negative breast cancers. Biomaterials 2013, 34, 10160–10171. [Google Scholar] [CrossRef]
- Zhao, D.; Zhang, H.; Yang, S.; He, W.; Luan, Y. Redox-sensitive mPEG-SS-PTX/TPGS mixed micelles: An efficient drug delivery system for overcoming multidrug resistance. Int. J. Pharm. 2016, 515, 281–292. [Google Scholar] [CrossRef]
- Singh, R.P.; Sharma, G.; Sonali; Agrawal, P.; Pandey, B.L.; Koch, B.; Muthu, M.S. Transferrin receptor targeted PLA-TPGS micelles improved efficacy and safety in docetaxel delivery. Int. J. Biol. Macromol. 2016, 83, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, S.; Ma, P.; Jiang, Y.; Cheng, K.; Yu, Y.; Jiang, N.; Miao, H.; Tang, Q.; Liu, F.; et al. Drug conjugate-based anticancer therapy—Current status and perspectives. Cancer Lett. 2023, 552, 215969. [Google Scholar] [CrossRef]
- Shi, D.; Beasock, D.; Fessler, A.; Szebeni, J.; Ljubimova, J.Y.; Afonin, K.A.; Dobrovolskaia, M.A. To PEGylate or not to PEGylate: Immunological properties of nanomedicine’s most popular component, polyethylene glycol and its alternatives. Adv. Drug Deliv. Rev. 2022, 180, 114079. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.H.; Guo, Z.; Jiang, D.; Zhou, X.; Lin, L.; Zhao, D.; Chen, M. Linear-like polypeptide-based micelle with pH-sensitive detachable PEG to deliver dimeric camptothecin for cancer therapy. Asian J. Pharm. Sci. 2023, 18, 100773. [Google Scholar] [CrossRef]
- Hu, J.; Obayemi, J.D.; Malatesta, K.; Košmrlj, A.; Soboyejo, W.O. Enhanced cellular uptake of LHRH-conjugated PEG-coated magnetite nanoparticles for specific targeting of triple negative breast cancer cells. Mater. Sci. Eng. C 2018, 88, 32–45. [Google Scholar] [CrossRef]
- Chen, Z.; Liang, Y.; Feng, X.; Liang, Y.; Shen, G.; Huang, H.; Chen, Z.; Yu, J.; Liu, H.; Lin, T.; et al. Vitamin-B12-conjugated PLGA-PEG nanoparticles incorporating miR-532-3p induce mitochondrial damage by targeting apoptosis repressor with caspase recruitment domain (ARC) on CD320-overexpressed gastric cancer. Mater. Sci. Eng. C 2021, 120, 111722. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Chen, J.; Xie, F.; Bao, W.; Xu, H.; Wang, H.; Xu, Y.; Du, Z. Herceptin-conjugated paclitaxel loaded PCL-PEG worm-like nanocrystal micelles for the combinatorial treatment of HER2-positive breast cancer. Biomaterials 2019, 222, 119420. [Google Scholar] [CrossRef]
- Mero, A.; Campisi, M. Hyaluronic acid bioconjugates for the delivery of bioactive molecules. Polymers 2014, 6, 346. [Google Scholar] [CrossRef]
- Dosio, F.; Arpicco, S.; Stella, B.; Fattal, E. Hyaluronic acid for anticancer drug and nucleic acid delivery. Adv. Drug Deliv. Rev. 2016, 97, 204–236. [Google Scholar] [CrossRef]
- Necas, J.; Bartosikava, L.; Brauner, P.; Kolar, J. Hyaluronic acid (hyaluronan): A review. Vet. Med. 2008, 53, 397–411. [Google Scholar] [CrossRef]
- Cowman, M.K.; Matsuoka, S. Experimental approaches to hyaluronan structure. Carbohydr. Res. 2005, 340, 791–809. [Google Scholar] [CrossRef]
- Toole, B.P.; Wight, T.N.; Tammi, M.I. Hyaluronan-cell interactions in cancer and vascular disease. J. Biol. Chem. 2002, 277, 4593–4596. [Google Scholar] [CrossRef]
- Liu, D.; Pearlman, E.; Diaconu, E.; Guo, K.; Mori, H.; Haqqi, T.; Markowitz, S.; Willson, J.; Sy, M.S. Expression of hyaluronidase by tumor cells induces angiogenesis in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 7832–7837. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Ghatak, S.; Toole, B.P. Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. J. Biol. Chem. 2005, 280, 20310–20315. [Google Scholar] [CrossRef] [PubMed]
- Qhattal, H.S.S.; Liu, X. Characterization of CD44-mediated cancer cell uptake and intracellular distribution of hyaluronan-grafted liposomes. Mol. Pharm. 2011, 8, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.; Oh, S.; Lee, K.-m.; Yoo, S.-a.; Shin, I. CD44 regulates cell proliferation, migration, and invasion via modulation of c-Src transcription in human breast cancer cells. Cell. Signal. 2015, 27, 1882–1894. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, V.; Vincent, I.R.; Gardner, H.; Chan, E.; Dhamko, H.; Jothy, S. CD44 regulates cell migration in human colon cancer cells via Lyn kinase and AKT phosphorylation. Exp. Mol. Pathol. 2007, 83, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Garay, J.; Piazuelo, M.B.; Majumdar, S.; Li, L.; Trillo-Tinoco, J.; Del Valle, L.; Schneider, B.G.; Delgado, A.G.; Wilson, K.T.; Correa, P.; et al. The homing receptor CD44 is involved in the progression of precancerous gastric lesions in patients infected with Helicobacter pylori and in development of mucous metaplasia in mice. Cancer Lett. 2016, 371, 90–98. [Google Scholar] [CrossRef]
- Takada, M.; Yamamoto, M.; Saitoh, Y. The significance of CD44 in human pancreatic cancer: II. The role of CD44 in human pancreatic adenocarcinoma invasion. Pancreas 1994, 9, 753–757. [Google Scholar] [CrossRef]
- Naor, D.; Wallach-Dayan, S.B.; Zahalka, M.A.; Sionov, R.V. Involvement of CD44, a molecule with a thousand faces, in cancer dissemination. Semin. Cancer Biol. 2008, 18, 260–267. [Google Scholar] [CrossRef]
- Fu, C.-P.; Cai, X.-Y.; Chen, S.-L.; Yu, H.-W.; Fang, Y.; Feng, X.-C.; Zhang, L.-M.; Li, C.-Y. Hyaluronic acid-based nanocarriers for anticancer drug delivery. Polymers 2023, 15, 2317. [Google Scholar] [CrossRef]
- Li, J.; Shin, G.H.; Chen, X.; Park, H.J. Modified curcumin with hyaluronic acid: Combination of pro-drug and nano-micelle strategy to address the curcumin challenge. Food Res. Int. 2015, 69, 202–208. [Google Scholar] [CrossRef]
- Krishnan, V.; Peng, K.; Sarode, A.; Prakash, S.; Zhao, Z.; Filippov, S.K.; Todorova, K.; Sell, B.R.; Lujano, O.; Bakre, S.; et al. Hyaluronic acid conjugates for topical treatment of skin cancer lesions. Sci. Adv. 2021, 7, abe6627. [Google Scholar] [CrossRef] [PubMed]
- Rosato, A.; Banzato, A.; De Luca, G.; Renier, D.; Bettella, F.; Pagano, C.; Esposito, G.; Zanovello, P.; Bassi, P. HYTAD1-p20: A new paclitaxel-hyaluronic acid hydrosoluble bioconjugate for treatment of superficial bladder cancer. Urol. Oncol. 2006, 24, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Xie, Y.; Bagby, T.R.; Cohen, M.S.; Forrest, M.L. Intralymphatic chemotherapy using a hyaluronan–cisplatin conjugate. J. Surg. Res. 2008, 147, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Thati, S.; Bagby, T.R.; Diab, H.-M.; Davies, N.M.; Cohen, M.S.; Forrest, M.L. Localized doxorubicin chemotherapy with a biopolymeric nanocarrier improves survival and reduces toxicity in xenografts of human breast cancer. J. Control. Release 2010, 146, 212–218. [Google Scholar] [CrossRef]
- Galer, C.E.; Sano, D.; Ghosh, S.C.; Hah, J.H.; Auzenne, E.; Hamir, A.N.; Myers, J.N.; Klostergaard, J. Hyaluronic acid-paclitaxel conjugate inhibits growth of human squamous cell carcinomas of the head and neck via a hyaluronic acid-mediated mechanism. Oral Oncol. 2011, 47, 1039–1047. [Google Scholar] [CrossRef]
- Mittapalli, R.K.; Liu, X.; Adkins, C.E.; Nounou, M.I.; Bohn, K.A.; Terrell, T.B.; Qhattal, H.S.; Geldenhuys, W.J.; Palmieri, D.; Steeg, P.S.; et al. Paclitaxel-hyaluronic nanoconjugates prolong overall survival in a preclinical brain metastases of breast cancer model. Mol. Cancer Ther. 2013, 12, 2389–2399. [Google Scholar] [CrossRef]
- Serafino, A.; Zonfrillo, M.; Andreola, F.; Psaila, R.; Mercuri, L.; Moroni, N.; Renier, D.; Campisi, M.; Secchieri, C.; Pierimarchi, P. CD44-targeting for antitumor drug delivery: A new SN-38-hyaluronan bioconjugate for locoregional treatment of peritoneal carcinomatosis. Curr. Cancer Drug Targets 2011, 11, 572–585. [Google Scholar] [CrossRef]
- Pang, X.; Lu, Z.; Du, H.; Yang, X.; Zhai, G. Hyaluronic acid-quercetin conjugate micelles: Synthesis, characterization, in vitro and in vivo evaluation. Colloids Surfaces. B Biointerfaces 2014, 123, 778–786. [Google Scholar] [CrossRef]
- Li, C.; Wallace, S. Polymer-drug conjugates: Recent development in clinical oncology. Adv. Drug Deliv. Rev. 2008, 60, 886–898. [Google Scholar] [CrossRef]
- Crucho, C.I.C.; Barros, M.T. Polymeric nanoparticles: A study on the preparation variables and characterization methods. Mater. Sci. Eng. C 2017, 80, 771–784. [Google Scholar] [CrossRef]
- Rodrigues de Azevedo, C.; von Stosch, M.; Costa, M.S.; Ramos, A.M.; Cardoso, M.M.; Danhier, F.; Préat, V.; Oliveira, R. Modeling of the burst release from PLGA micro- and nanoparticles as function of physicochemical parameters and formulation characteristics. Int. J. Pharm. 2017, 532, 229–240. [Google Scholar] [CrossRef]
- Cesar, A.L.A.; Abrantes, F.A.; Farah, L.; Castilho, R.O.; Cardoso, V.; Fernandes, S.O.; Araújo, I.D.; Faraco, A.A.G. New mesalamine polymeric conjugate for controlled release: Preparation, characterization and biodistribution study. Eur. J. Pharm. Sci. 2018, 111, 57–64. [Google Scholar] [CrossRef]
- Suksiriworapong, J.; Taresco, V.; Ivanov, D.P.; Styliari, I.D.; Sakchaisri, K.; Junyaprasert, V.B.; Garnett, M.C. Synthesis and properties of a biodegradable polymer-drug conjugate: Methotrexate-poly(glycerol adipate). Colloids Surf. B Biointerfaces 2018, 167, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.; Gaspar, H.B. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID). Biol. Targets Ther. 2009, 3, 349–358. [Google Scholar]
- Lainka, E.; Hershfield, M.S.; Santisteban, I.; Bali, P.; Seibt, A.; Neubert, J.; Friedrich, W.; Niehues, T. polyethylene glycol-conjugated adenosine deaminase (ADA) therapy provides temporary immune reconstitution to a child with delayed-onset ADA deficiency. Clin. Diagn. Lab. Immunol. 2005, 12, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.A.; Kularatne, S.A.; Hakenjos, J.; Carron, J.D.; Henne, K.L. Synthesis and activity of a folate targeted monodisperse PEG camptothecin conjugate. Bioorganic Med. Chem. Lett. 2013, 23, 5810–5813. [Google Scholar] [CrossRef] [PubMed]
- Thummarati, P.; Suksiriworapong, J.; Sakchaisri, K.; Junyaprasert, V.B. Effect of chemical linkers of curcumin conjugated hyaluronic acid on nanoparticle properties and in vitro performances in various cancer cells. J. Drug Deliv. Sci. Technol. 2021, 61, 102323. [Google Scholar] [CrossRef]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block copolymer micelles in nanomedicine applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef]
- Tang, M.; Zhou, M.; Huang, Y.; Zhong, J.; Zhou, Z.; Luo, K. Dual-sensitive and biodegradable core-crosslinked HPMA copolymer–doxorubicin conjugate-based nanoparticles for cancer therapy. Polym. Chem. 2017, 8, 2370–2380. [Google Scholar] [CrossRef]
- Dey, S.; Sreenivasan, K. Conjugation of curcumin onto alginate enhances aqueous solubility and stability of curcumin. Carbohydr. Polym. 2014, 99, 499–507. [Google Scholar] [CrossRef]
- Ding, C.; Li, Z. A review of drug release mechanisms from nanocarrier systems. Mater. Sci. Eng. C 2017, 76, 1440–1453. [Google Scholar] [CrossRef]
- Pang, X.; Jiang, Y.; Xiao, Q.; Leung, A.W.; Hua, H.; Xu, C. pH-responsive polymer–drug conjugates: Design and progress. J. Control. Release 2016, 222, 116–129. [Google Scholar] [CrossRef]
- Yang, T.; Feng, W.; Hu, C.; Lv, Z.; Wei, H.; Jiang, J.; Liu, S.; Zhao, Q. Manganese porphyrin-incorporated conjugated polymer nanoparticles for T1-enhanced magnetic resonance and fluorescent imaging. Inorg. Chim. Acta 2017, 466, 604–611. [Google Scholar] [CrossRef]
- Gupta, P.; Vermani, K.; Garg, S. Hydrogels: From controlled release to pH-responsive drug delivery. Drug Discov. Today 2002, 7, 569–579. [Google Scholar] [CrossRef]
- Kost, J.; Langer, R. Responsive polymeric delivery systems. Adv. Drug Deliv. Rev. 2001, 46, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Yamamoto, S.; Nakaki, R.; Shimamura, T.; Hamakubo, T.; Sakai, J.; Kodama, T.; Yoshida, T.; Aburatani, H.; Osawa, T. Extracellular acidic pH activates the sterol regulatory element-binding protein 2 to promote tumor progression. Cell Rep. 2017, 18, 2228–2242. [Google Scholar] [CrossRef]
- Koltai, T. Cancer: Fundamentals behind pH targeting and the double-edged approach. OncoTargets Ther. 2016, 9, 6343–6360. [Google Scholar] [CrossRef]
- Jiang, T.; Li, Y.-M.; Lv, Y.; Cheng, Y.-J.; He, F.; Zhuo, R.-X. Amphiphilic polycarbonate conjugates of doxorubicin with pH-sensitive hydrazone linker for controlled release. Colloids Surf. B Biointerfaces 2013, 111, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.; Cui, H. Peptide–drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Gu, L.; Sun, Y.; Wang, S.; Zhang, X.; Zhu, J.; Sun, B. A series of enzyme-controlled-release polymer-platinum-based drug conjugates for the treatment of gastric cancer. Eur. Polym. J. 2017, 92, 105–116. [Google Scholar] [CrossRef]
- Zhang, C.; Pan, D.; Li, J.; Hu, J.; Bains, A.; Guys, N.; Zhu, H.; Li, X.; Luo, K.; Gong, Q.; et al. Enzyme-responsive peptide dendrimer-gemcitabine conjugate as a controlled-release drug delivery vehicle with enhanced antitumor efficacy. Acta Biomater. 2017, 55, 153–162. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Jain, R.K. Strategies for advancing cancer nanomedicine. Nat. Mater. 2013, 12, 958. [Google Scholar] [CrossRef]
- Shivhare, K.; Garg, C.; Priyam, A.; Gupta, A.; Sharma, A.K.; Kumar, P. Enzyme sensitive smart inulin-dehydropeptide conjugate self-assembles into nanostructures useful for targeted delivery of ornidazole. Int. J. Biol. Macromol. 2018, 106, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.; Tan, F.E.; Langer, R. Synthesis and characterization of dextran−peptide−methotrexate conjugates for tumor targeting via mediation by matrix metalloproteinase II and matrix metalloproteinase IX. Bioconjugate Chem. 2004, 15, 931–941. [Google Scholar] [CrossRef]
- Rejmanová, P.; Kopeček, J.; Pohl, J.; Baudyš, M.; Kostka, V. Polymers containing enzymatically degradable bonds, 8*. Degradation of oligopeptide sequences in N-(2-hydroxypropyl)methacrylamide copolymers by bovine spleen cathepsin B. Macomolecular Chem. Phys. 1983, 184, 2009–2020. [Google Scholar]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, P.; Cheetham, A.G.; Moon, J.H.; Moxley, J.W.; Lin, Y.-a.; Cui, H. Controlled release of free doxorubicin from peptide–drug conjugates by drug loading. J. Control. Release 2014, 191, 123–130. [Google Scholar] [CrossRef]
- Chandran, S.S.; Nan, A.; Rosen, D.M.; Ghandehari, H.; Denmeade, S.R. A prostate-specific antigen activated N-(2-hydroxypropyl) methacrylamide copolymer prodrug as dual-targeted therapy for prostate cancer. Mol. Cancer Ther. 2007, 6, 2928–2937. [Google Scholar] [CrossRef]
- Akimoto, J.; Nakayama, M.; Okano, T. Temperature-responsive polymeric micelles for optimizing drug targeting to solid tumors. J. Control. Release 2014, 193, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Campora, S.; Mohsen, R.; Passaro, D.; Samir, H.; Ashraf, H.; Al-Mofty, S.E.; Diab, A.A.; El-Sherbiny, I.M.; Snowden, M.J.; Ghersi, G. Functionalized Poly(N-isopropylacrylamide)-Based Microgels in Tumor Targeting and Drug Delivery. Gels 2021, 7, 203. [Google Scholar] [CrossRef] [PubMed]
- Farjadian, F.R.S.; Naeimi, M.; Ghasemi, S.; Mohammadi-Samani, S.; Welland, M.E.; Tayebi, L. Temperature and pH-responsive nano-hydrogel drug delivery system based on lysine-modified poly(vinylcaprolactam). Int. J. Nanomed. 2019, 14, 690115. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.M.; Suneetha, M.; Kumar, D.V.; Kim, H.J.; Seok, Y.J.; Han, S.S. Dual responsive poly(vinyl caprolactam)-based nanogels for tunable intracellular doxorubicin delivery in cancer cells. Pharmaceutics 2022, 14, 852. [Google Scholar] [CrossRef]
- Li, G.; Li, Y.; Tang, Y.; Zhang, Y.; Zhang, Y.; Yin, T.; Xu, H.; Cai, C.; Tang, X. Hydroxyethyl starch conjugates for improving the stability, pharmacokinetic behavior and antitumor activity of 10-hydroxy camptothecin. Int. J. Pharm. 2014, 471, 234–244. [Google Scholar] [CrossRef]
- Omar, R.; Bardoogo, Y.L.; Corem-Salkmon, E.; Mizrahi, B. Amphiphilic star PEG-camptothecin conjugates for intracellular targeting. J. Control. Release 2017, 257, 76–83. [Google Scholar] [CrossRef]
- Plichta, A.; Kowalczyk, S.; Olędzka, E.; Sobczak, M.; Strawski, M. Effect of structural factors on release profiles of camptothecin from block copolymer conjugates with high load of drug. Int. J. Pharm. 2018, 538, 231–242. [Google Scholar] [CrossRef]
- Davis, M.E. Design and development of IT-101, a cyclodextrin-containing polymer conjugate of camptothecin. Adv. Drug Deliv. Rev. 2009, 61, 1189–1192. [Google Scholar] [CrossRef]
- Dal Pozzo, A.; Ni, M.-H.; Esposito, E.; Dallavalle, S.; Musso, L.; Bargiotti, A.; Pisano, C.; Vesci, L.; Bucci, F.; Castorina, M.; et al. Novel tumor-targeted RGD peptide–camptothecin conjugates: Synthesis and biological evaluation. Bioorganic Med. Chem. 2010, 18, 64–72. [Google Scholar] [CrossRef]
- Brannon-Peppas, L.; Blanchette, J.O. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev 2004, 56, 1649–1659. [Google Scholar] [CrossRef]
- Kou, L.; Sun, J.; Zhai, Y.; He, Z. The endocytosis and intracellular fate of nanomedicines: Implication for rational design. Asian J. Pharm. Sci. 2013, 8, 1–10. [Google Scholar] [CrossRef]
- Sun, L.; Wu, Q.; Peng, F.; Liu, L.; Gong, C. Strategies of polymeric nanoparticles for enhanced internalization in cancer therapy. Colloids Surf. B Biointerfaces 2015, 135, 56–72. [Google Scholar] [CrossRef]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef]
- Chen, Z. Small-molecule delivery by nanoparticles for anticancer therapy. Trends Mol. Med. 2010, 16, 594–602. [Google Scholar] [CrossRef]
- Maeda, H. SMANCS and polymer-conjugated macromolecular drugs: Advantages in cancer chemotherapy. Adv. Drug Deliv. Rev. 2001, 46, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chen, D.; Li, L.; Sun, K. Charge reversal nano-systems for tumor therapy. J. Nanobiotechnol. 2022, 20, 31. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Leroux, J.-C. The journey of a drug-carrier in the body: An anatomo-physiological perspective. J. Control. Release 2012, 161, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Bharate, G.Y.; Daruwalla, J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur. J. Pharm. Biopharm. 2009, 71, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Li, Y.; Luo, J.; Lee, J.S.; Xiao, W.; Gonik, A.M.; Agarwal, R.G.; Lam, K.S. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials 2011, 32, 3435–3446. [Google Scholar] [CrossRef]
- Nagpure, G.; Rb Singh, K.; Singh, J.; Singh, R.P. Chapter 10—Passive and Active Targeted drug Delivery Strategies. In Nanotechnology for Drug Delivery and Pharmaceuticals; Pratap Singh, R., Rb Singh, K., Singh, J., Adetunji, C.O., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 225–234. [Google Scholar]
- Zamani, M.; Rostamizadeh, K.; Kheiri Manjili, H.; Danafar, H. In vitro and in vivo biocompatibility study of folate-lysine-PEG-PCL as nanocarrier for targeted breast cancer drug delivery. Eur. Polym. J. 2018, 103, 260–270. [Google Scholar] [CrossRef]
- Huang, G.; Huang, H. Hyaluronic acid-based biopharmaceutical delivery and tumor-targeted drug delivery system. J. Control. Release 2018, 278, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-H.; Jeong, G.-W.; Nah, J.-W. Preparation and anticancer effect of transferrin-modified pH-sensitive polymeric drug nanoparticle for targeted cancer therapy. J. Ind. Eng. Chem. 2017, 54, 298–303. [Google Scholar] [CrossRef]
- Isaacson, K.J.; Martin Jensen, M.; Subrahmanyam, N.B.; Ghandehari, H. Matrix-metalloproteinases as targets for controlled delivery in cancer: An analysis of upregulation and expression. J. Control. Release 2017, 259, 62–75. [Google Scholar] [CrossRef]
- Vogus, D.R.; Evans, M.A.; Pusuluri, A.; Barajas, A.; Zhang, M.; Krishnan, V.; Nowak, M.; Menegatti, S.; Helgeson, M.E.; Squires, T.M.; et al. A hyaluronic acid conjugate engineered to synergistically and sequentially deliver gemcitabine and doxorubicin to treat triple negative breast cancer. J. Control. Release 2017, 267, 191–202. [Google Scholar] [CrossRef]
- Xu, Z.; Zheng, W.; Yin, Z. Synthesis and optimization of a bifunctional hyaluronan-based camptothecin prodrug. Arch. Pharm. 2014, 347, 240–246. [Google Scholar] [CrossRef]
- Xie, Y.; Aillon, K.L.; Cai, S.; Christian, J.M.; Davies, N.M.; Berkland, C.J.; Forrest, M.L. Pulmonary delivery of cisplatin-hyaluronan conjugates via endotracheal instillation for the treatment of lung cancer. Int. J. Pharm. 2010, 392, 156–163. [Google Scholar] [CrossRef]
- Reynolds, J.G.; Geretti, E.; Hendriks, B.S.; Lee, H.; Leonard, S.C.; Klinz, S.G.; Noble, C.O.; Lücker, P.B.; Zandstra, P.W.; Drummond, D.C.; et al. HER2-targeted liposomal doxorubicin displays enhanced anti-tumorigenic effects without associated cardiotoxicity. Toxicol. Appl. Pharmacol. 2012, 262, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, P.; Abnous, K.; Taghdisi, S.M.; Zahiri, M.; Ramezani, M.; Alibolandi, M. Targeted MMP-2 responsive chimeric polymersomes for therapy against colorectal cancer. Colloids Surf. B Biointerfaces 2020, 193, 111135. [Google Scholar] [CrossRef]
- Mi, Y.; Wolfram, J.; Mu, C.; Liu, X.; Blanco, E.; Shen, H.; Ferrari, M. Enzyme-responsive multistage vector for drug delivery to tumor tissue. Pharmacol. Res. 2016, 113, 92–99. [Google Scholar] [CrossRef]
- Dai, Z.; Yao, Q.; Zhu, L. MMP2-sensitive PEG–Lipid copolymers: A new type of tumor-targeted P-glycoprotein inhibitor. ACS Appl. Mater. Interfaces 2016, 8, 12661–12673. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, B.; Meng, X.; She, P.; Zhang, P.; Cao, Y.; Zhang, X. Preparation of dual-drug conjugated polymeric micelles with synergistic anti-cancer efficacy in vitro. J. Drug Deliv. Sci. Technol. 2018, 43, 388–396. [Google Scholar] [CrossRef]
- Sarika, P.R.; James, N.R.; Nishna, N.; Anil Kumar, P.R.; Raj, D.K. Galactosylated pullulan–curcumin conjugate micelles for site specific anticancer activity to hepatocarcinoma cells. Colloids Surf. B Biointerfaces 2015, 133, 347–355. [Google Scholar] [CrossRef]
- Hoang, B.; Ernsting, M.J.; Tang, W.-H.S.; Bteich, J.; Undzys, E.; Kiyota, T.; Li, S.-D. Cabazitaxel-conjugated nanoparticles for docetaxel-resistant and bone metastatic prostate cancer. Cancer Lett. 2017, 410, 169–179. [Google Scholar] [CrossRef] [PubMed]
- van Haandel, L.; Stobaugh, J.F. Bioanalytical method development for a generation 5 polyamidoamine folic acid methotrexate conjugated nanoparticle. Anal. Bioanal. Chem. 2010, 397, 1841–1852. [Google Scholar] [CrossRef]
- Badkas, A.; Frank, E.; Zhou, Z.; Jafari, M.; Chandra, H.; Sriram, V.; Lee, J.-Y.; Yadav, J.S. Modulation of in vitro phagocytic uptake and immunogenicity potential of modified Herceptin®-conjugated PLGA-PEG nanoparticles for drug delivery. Colloids Surf. B Biointerfaces 2018, 162, 271–278. [Google Scholar] [CrossRef] [PubMed]











| Polymer Compositions | Grafting Ligand | Drug | Disease | Application | Ref. |
|---|---|---|---|---|---|
| Folic acid-PCL-PEG | Folic acid | MTX | Breast cancer | Enhanced cytotoxicity and specificity | [12,50] |
| Oleic acid-PEG-b-PCL | Oleic acid | Curcumin (CUR) | Brain cancer | Enhanced accumulation in the brain | [52] |
| Folic acid-(P(CL)2-PEG | Folic acid | MTX | Breast cancer | Enhanced cytotoxicity and specificity | [12,50] |
| PCL-TPGS | - | Quercetin (QCT) | Breast cancer | Enhanced drug loading capacity Sustained drug release | [44] |
| Bi(mPEG-SeSe)-PCL | - | DOX | Skin cancer | Enhanced cytotoxicity and specificity | [53] |
| Polymer Compositions | Grafting Ligand | Drug | Application | Ref. |
|---|---|---|---|---|
| TPGS | DOX | Increased drug stability Enhanced cellular uptake and efficacy Reduced side effects in vivo | [70] | |
| TPGS | CIS | Enhanced the efficacy Presented neuroprotective effect | [60] | |
| TPGS | GEM | Improved cytotoxicity | [71] | |
| TPGS | DTX Cetuximab (Cmab) | Achieved synergistic effects for multidrug resistance Enhanced the efficacy | [72] | |
| mPEG-paclitaxel/TPGS | PTX | Achieved synergistic effects for multidrug resistance Enhanced cellular uptake. Enhanced the efficacy | [73] | |
| TPGS-b-PCL/Pluronic P123 | Anti-GPC3 antibody | Sorafenib (Sf) | Enhanced cellular uptake and cytotoxicity in liver cancer | [68] |
| PLA-TPGS | Transferrin | DTX | Improved pharmacokinetic profile Enhanced cytotoxicity and efficiency in vivo | [74] |
| TPGS/TPGS | Folic acid | MTX | Enhanced the targeted drug delivery | [61] |
| MW of HA (kDa) | Drug | Administration Route * | Disease | Tumor Model | Ref. |
|---|---|---|---|---|---|
| 200 | PTX | i.p. and i.v.c | Ovarian cancer, bladder cancer | OVCAR-3, SKOV-3, Phase II clinical trial | [97] |
| 40 | PTX | i.v. | Squamous cell carcinoma of the head and neck | OSC-19, NH5 | [100] |
| 5 | PTX | i.v. | Brain metastasis, breast cancer | 231 Br | [101] |
| 35 | DOX | s.c. | Breast cancer | MDA-MB-468LN | [99] |
| 200 | CPT | i.p. | Peritoneal cancer | HT-29, MKN-45, OE-21, DHD/K21/Trb | [102] |
| 35 | CIS | s.c. | Breast cancer | MCF-7, MDA-MB-231 | [98] |
| 10 | QCT | i.v. | Hepatoma | H22 | [103] |
| 11 | GEM/CUR | i.v. | Pancreatic cancer, colon cancer, lung cancer | PANC-1 Caco-2, HCT116 A549 | [34] |
| Polymers | Drugs | %DL * (%w/w) | Solubility in Water | Application | Ref. | |
|---|---|---|---|---|---|---|
| Conventional | PDCs | |||||
| HA | CUR | 1.3+0.31 | 0.27 µg/mL | 7.5 mg/mL | Improved stability | [115] |
| PEG | PTX | 60.3 | <2 μg/mL | 3665 μg/mL | Human cervical carcinoma | [11] |
| Polymer Compositions | Drugs | Specific Enzymes | Stage | Application | Ref. |
|---|---|---|---|---|---|
| HPMA-Gly-Phe-Leu-Gly | - | Cathepsin B | In vitro | Increased stability in plasma | [130] |
| Brentuximab vedotin-Val-Cit-PABC | Monomethyl auristatin E | Cathepsin B | FDA approval | Used for Hodkin lymphoma | [131] |
| PEG-Gly-Cys-Gly-Ala-Ala-Asn-Leu-Glu | CIS | Legumain | In vitro | Increased drug stability in plasma; enhanced gastric cancer therapy | [125] |
| PEG-Gly-Phe-Leu-Gly | GEM | Cathepsin B | In vitro and in vivo | Increased drug stability in plasma; increased antitumor activity in breast cancer, but reduced side effects to normal tissues | [126] |
| NTD-Gly-Phe-Leu-Gly | DOX | Cathepsin B | In vitro | Increased stability in plasma; enhanced drug accumulation in liver cancer cells | [132] |
| Dextran-Pro-Val-Gly-Leu-Ile-Gly | MTX | MMP-2/MMP-9 | In vitro | Increased stability in plasma; enhanced drug accumulation in fibrosarcoma cell line and liver cancer cells | [129] |
| HPMA-morpholinocarbonyl-Ser-Ser-Lys-Tyr-Gln-Leu | 12-aminododecanoyl thapsigargin | Cathepsin B | In vitro and in vivo | Enhanced drug accumulation in prostate cancer cells | [133] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Junyaprasert, V.B.; Thummarati, P. Innovative Design of Targeted Nanoparticles: Polymer–Drug Conjugates for Enhanced Cancer Therapy. Pharmaceutics 2023, 15, 2216. https://doi.org/10.3390/pharmaceutics15092216
Junyaprasert VB, Thummarati P. Innovative Design of Targeted Nanoparticles: Polymer–Drug Conjugates for Enhanced Cancer Therapy. Pharmaceutics. 2023; 15(9):2216. https://doi.org/10.3390/pharmaceutics15092216
Chicago/Turabian StyleJunyaprasert, Varaporn Buraphacheep, and Parichart Thummarati. 2023. "Innovative Design of Targeted Nanoparticles: Polymer–Drug Conjugates for Enhanced Cancer Therapy" Pharmaceutics 15, no. 9: 2216. https://doi.org/10.3390/pharmaceutics15092216
APA StyleJunyaprasert, V. B., & Thummarati, P. (2023). Innovative Design of Targeted Nanoparticles: Polymer–Drug Conjugates for Enhanced Cancer Therapy. Pharmaceutics, 15(9), 2216. https://doi.org/10.3390/pharmaceutics15092216