
Prenatal Exposure to BPA: The Effects on Hepatic Lipid Metabolism in Male and Female Rat Fetuses

 ,
,  , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals and Treatment
2.3. Measurement of Triglycerides and Cholesterol, HDL, and LDL Content in Serum and Liver Samples
2.4. Measurement of Tumor Necrosis Factor Alpha (TNF-α) in Liver Samples
2.5. Total Lysate and Membrane Preparation for Western Blot Analysis
2.6. Immunoblotting
2.7. In Vitro HMGCR Degradation Assay
2.8. Statistical Analysis
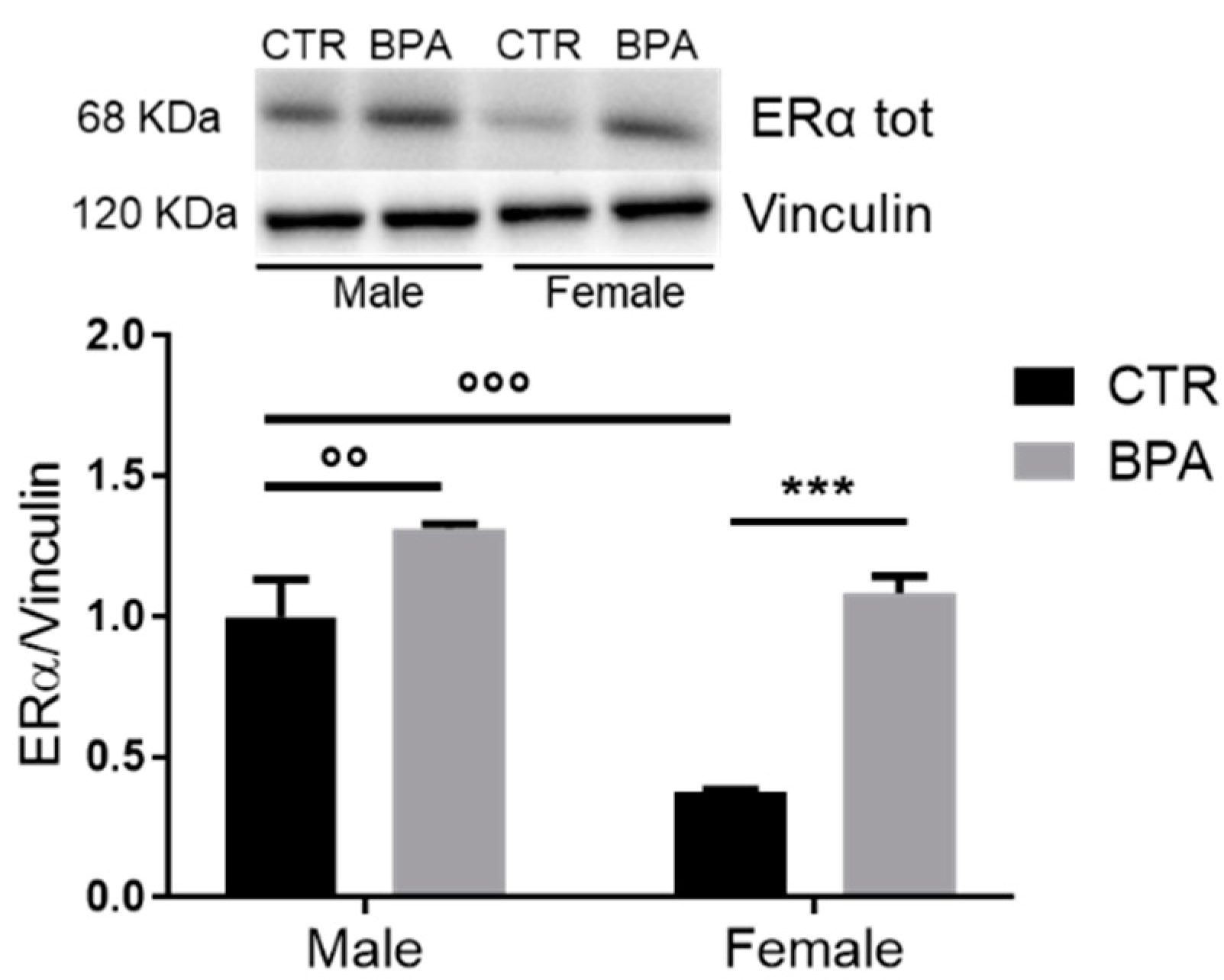
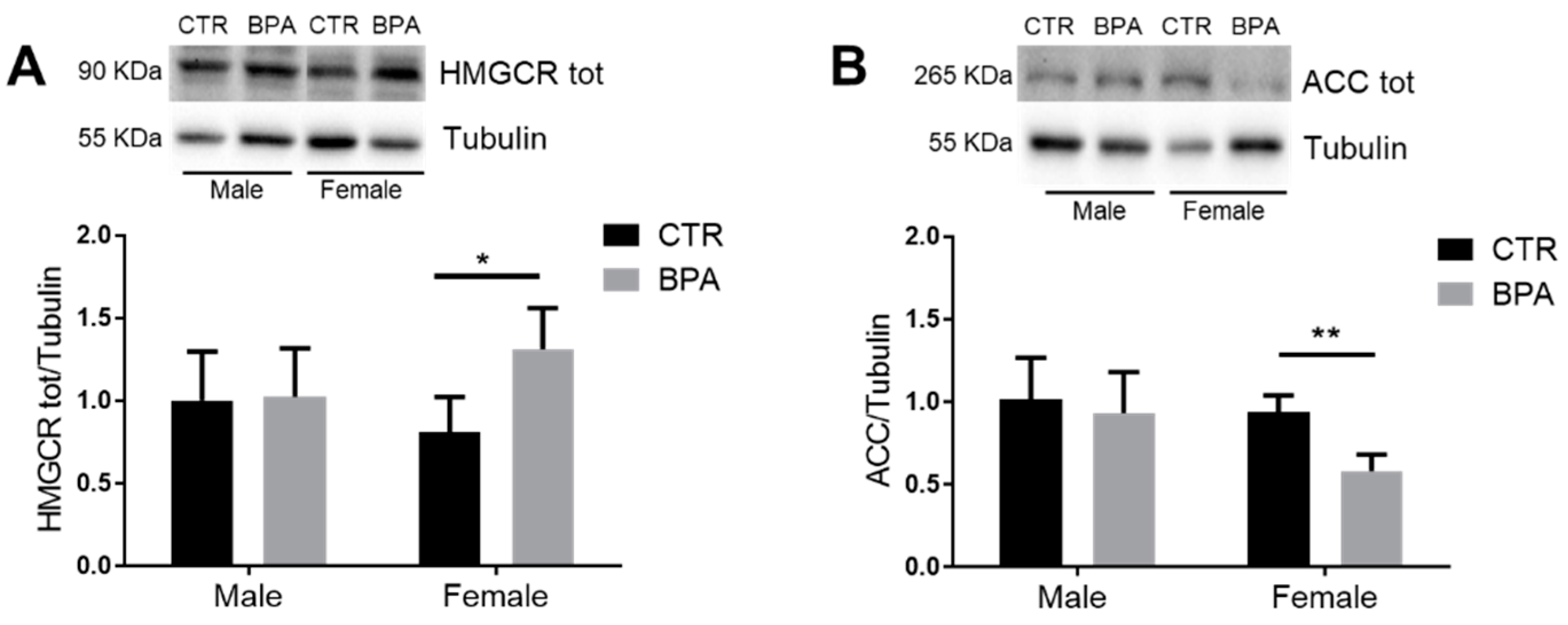
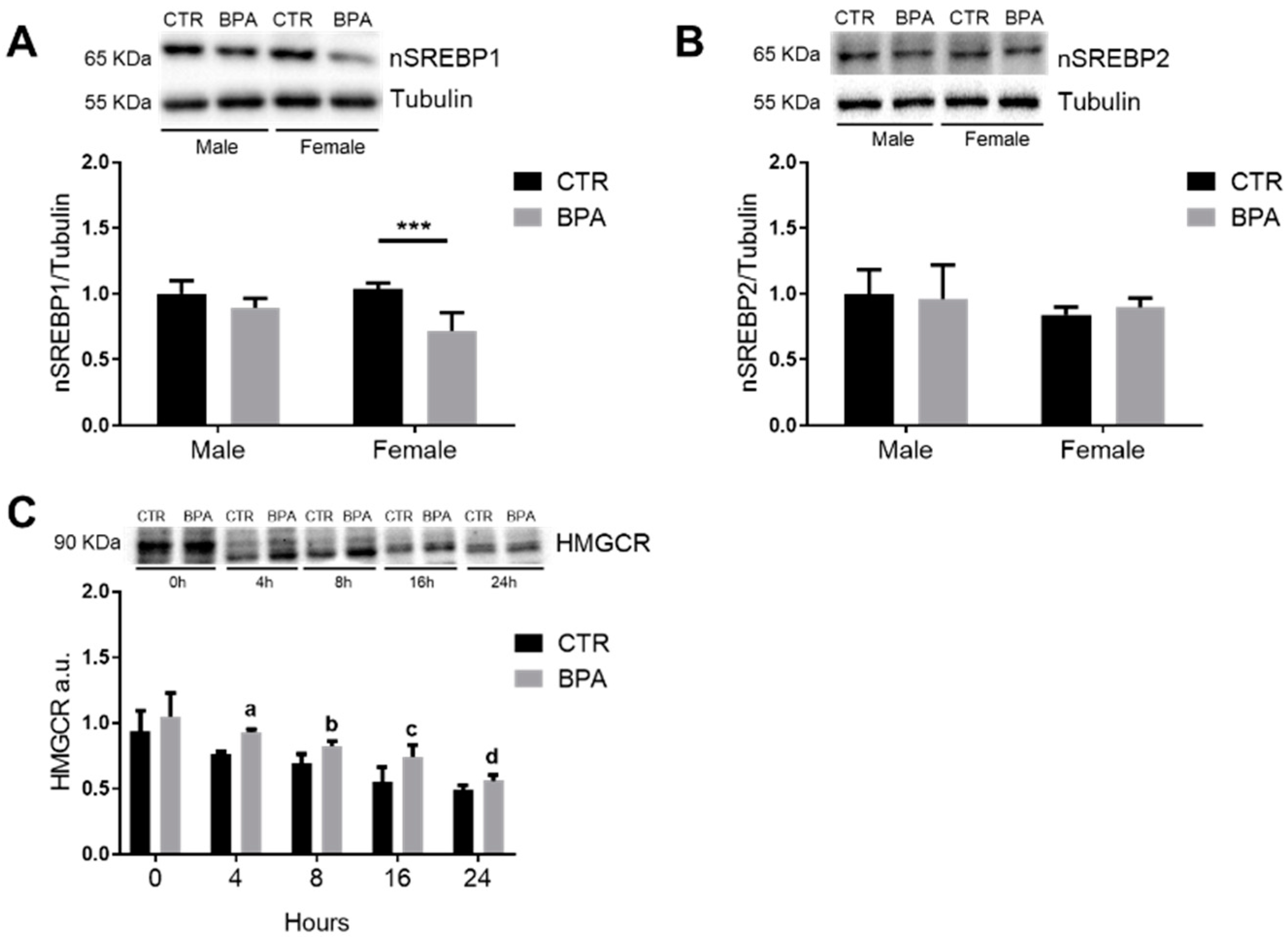
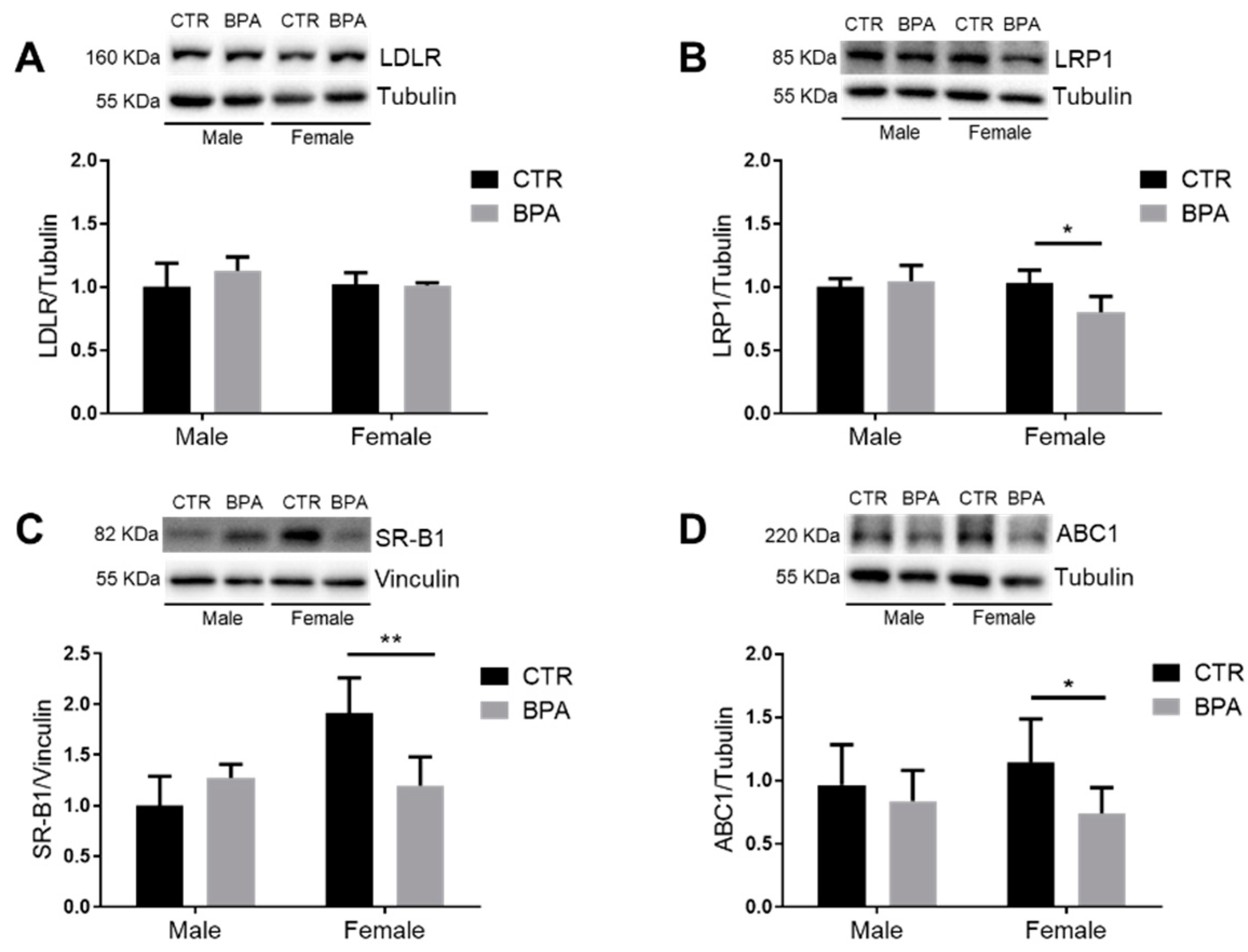
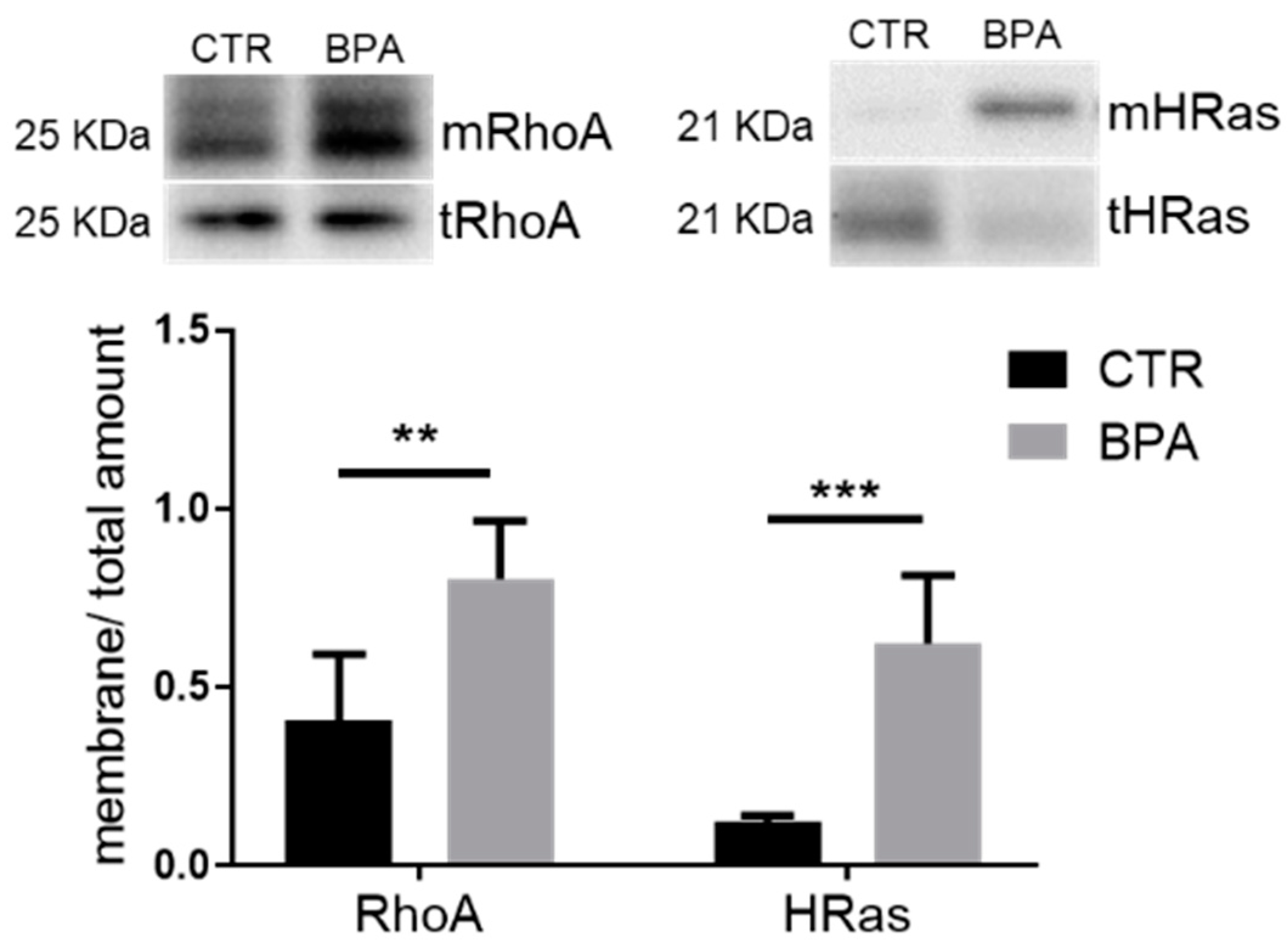
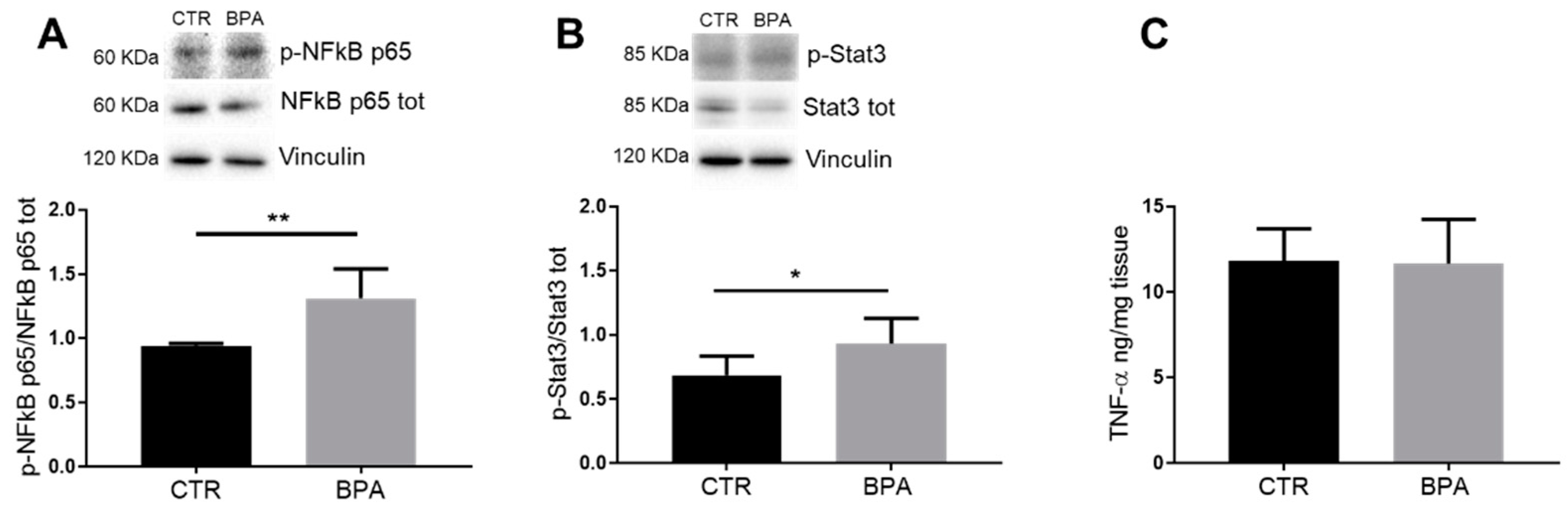
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murata, M.; Kang, J.-H. Bisphenol A (BPA) and cell signaling pathways. Biotechnol. Adv. 2018, 36, 311–327. [Google Scholar] [CrossRef]
- Geens, T.; Goeyens, L.; Covaci, A. Are potential sources for human exposure to bisphenol-A overlooked? Int. J. Hyg. Environ. Health 2011, 214, 339–347. [Google Scholar] [CrossRef]
- Geens, T.; Aerts, D.; Berthot, C.; Bourguignon, J.-P.; Goeyens, L.; Lecomte, P.; Maghuin-Rogister, G.; Pironnet, A.-M.; Pussemier, L.; Scippo, M.-L.; et al. A review of dietary and non-dietary exposure to bisphenol-A. Food Chem. Toxicol. 2012, 50, 3725–3740. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-H.; Kondo, F.; Katayama, Y. Human exposure to bisphenol A. Toxicology 2006, 226, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Covaci, A.; Hond, E.D.; Geens, T.; Govarts, E.; Koppen, G.; Frederiksen, H.; Knudsen, L.E.; Mørck, T.A.; Gutleb, A.C.; Guignard, C.; et al. Urinary BPA measurements in children and mothers from six European member states: Overall results and determinants of exposure. Environ. Res. 2015, 141, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Teeguarden, J.G.; Twaddle, N.C.; Churchwell, M.I.; Doerge, D.R. Urine and serum biomonitoring of exposure to environmental estrogens I: Bisphenol A in pregnant women. Food Chem. Toxicol. 2016, 92, 129–142. [Google Scholar] [CrossRef]
- Gerona, R.R.; Woodruff, T.J.; Dickenson, C.A.; Pan, J.; Schwartz, J.M.; Sen, S.; Friesen, M.W.; Fujimoto, V.Y.; Hunt, P.A. Bisphenol-A (BPA), BPA Glucuronide, and BPA Sulfate in Midgestation Umbilical Cord Serum in a Northern and Central California Population. Environ. Sci. Technol. 2013, 47, 12477–12485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.-L.; Popovic, S.; Arbuckle, T.E.; Fraser, W.D. Determination of free and total bisphenol A in human milk samples from Canadian women using a sensitive and selective GC-MS method. Food Addit. Contam. Part A 2014, 32, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Kirkley, A.G.; Sargis, R.M. Environmental endocrine disruption of energy metabolism and cardiovascular risk. Curr. Diabetes Rep. 2014, 14, 494. [Google Scholar] [CrossRef] [Green Version]
- Grasselli, E.; Cortese, K.; Voci, A.; Vergani, L.; Fabbri, R.; Barmo, C.; Gallo, G.; Canesi, L. Direct effects of Bisphenol A on lipid homeostasis in rat hepatoma cells. Chemosphere 2013, 91, 1123–1129. [Google Scholar] [CrossRef]
- Wang, T.; Li, M.; Chen, B.; Xu, M.; Xu, Y.; Huang, Y.; Lu, J.; Chen, Y.; Wang, W.; Li, X.; et al. Urinary Bisphenol A (BPA) Concentration Associates with Obesity and Insulin Resistance. J. Clin. Endocrinol. Metab. 2012, 97, E223–E227. [Google Scholar] [CrossRef] [Green Version]
- Rochester, J.R. Bisphenol A and human health: A review of the literature. Reprod. Toxicol. 2013, 42, 132–155. [Google Scholar] [CrossRef] [PubMed]
- Rönn, M.; Kullberg, J.; Karlsson, H.; Berglund, J.; Malmberg, F.; Örberg, J.; Lind, L.; Ahlström, H.; Lind, P.M. Bisphenol A exposure increases liver fat in juvenile fructose-fed Fischer 344 rats. Toxicology 2013, 303, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghaddam, H.S.; Samarghandian, S.; Farkhondeh, T. Effect of bisphenol A on blood glucose, lipid profile and oxidative stress indices in adult male mice. Toxicol. Mech. Methods 2015, 25, 507–513. [Google Scholar] [CrossRef] [PubMed]
- García-Arevalo, M.; Magdalena, P.A.; Dos Santos, J.R.; Quesada, I.; Carneiro, E.M.; Nadal, A. Exposure to Bisphenol-A during Pregnancy Partially Mimics the Effects of a High-Fat Diet Altering Glucose Homeostasis and Gene Expression in Adult Male Mice. PLoS ONE 2014, 9, e100214. [Google Scholar] [CrossRef] [Green Version]
- Tonini, C.; Segatto, M.; Gagliardi, S.; Bertoli, S.; Leone, A.; Barberio, L.; Mandalà, M.; Pallottini, V. Maternal Dietary Exposure to Low-Dose Bisphenol A Affects Metabolic and Signaling Pathways in the Brain of Rat Fetuses. Nutrients 2020, 12, 1448. [Google Scholar] [CrossRef]
- Donaldson, W.E. Regulation of Fatty Acid Synthesis. Fed. Proc. 1979, 38, 2617–2621. [Google Scholar]
- Brown, M.S.; Goldstein, J.L. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J. Lipid Res. 1980, 21, 505–517. [Google Scholar] [CrossRef]
- Espenshade, P.J.; Hughes, A.L. Regulation of Sterol Synthesis in Eukaryotes. Annu. Rev. Genet. 2007, 41, 401–427. [Google Scholar] [CrossRef]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochim. 2004, 86, 839–848. [Google Scholar] [CrossRef]
- Go, G.-W.; Mani, A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J. Biol. Med. 2012, 85, 19–28. [Google Scholar]
- Shen, W.-J.; Azhar, S.; Kraemer, F.B. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu. Rev. Physiol. 2018, 80, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Zaulet, M.; Kevorkian, S.E.M.; Dinescu, S.; Cotoraci, C.; Suciu, M.; Herman, H.; Buburuzan, L.; Badulescu, L.A.; Ardelean, A.; Hermenean, A. Protective effects of silymarin against bisphenol A-induced hepatotoxicity in mouse liver. Exp. Ther. Med. 2017, 13, 821–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirozzi, C.; Lama, A.; Annunziata, C.; Cavaliere, G.; Ruiz-Fernandez, C.; Monnolo, A.; Comella, F.; Gualillo, O.; Stornaiuolo, M.; Mollica, M.P.; et al. Oral Bisphenol A Worsens Liver Immune-Metabolic and Mitochondrial Dysfunction Induced by High-Fat Diet in Adult Mice: Cross-Talk between Oxidative Stress and Inflammasome Pathway. Antioxidants 2020, 9, 1201. [Google Scholar] [CrossRef]
- Moon, M.K.; Kim, M.J.; Jung, I.K.; Koo, Y.D.; Ann, H.Y.; Lee, K.J.; Kim, S.H.; Yoon, Y.C.; Cho, B.-J.; Park, K.S.; et al. Bisphenol A Impairs Mitochondrial Function in the Liver at Doses below the No Observed Adverse Effect Level. J. Korean Med. Sci. 2012, 27, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.H.S.; Andrade, J.M.O.; Fernandes, L.R.; Sinisterra, R.D.; Sousa, F.B.; Feltenberger, J.D.; Alvarez-Leite, J.I.; Santos, R.A.S. Oral Angiotensin-(1–7) prevented obesity and hepatic inflammation by inhibition of resistin/TLR4/MAPK/NF-κB in rats fed with high-fat diet. Pepttides 2013, 46, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Materials, E.E.P.O.F.C. Scientific Opinion on the risks to public health related to the presence of bisphenol A (BPA) in foodstuffs. EFSA J. 2015, 13. [Google Scholar] [CrossRef]
- Spagnuolo, M.S.; Pallottini, V.; Mazzoli, A.; Iannotta, L.; Tonini, C.; Morone, B.; Ståhlman, M.; Crescenzo, R.; Strazzullo, M.; Iossa, S.; et al. A Short-Term Western Diet Impairs Cholesterol Homeostasis and Key Players of Beta Amyloid Metabolism in Brain of Middle Aged Rats. Mol. Nutr. Food Res. 2020, 64, e2000541. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Acconcia, F.; Pallottini, V.; Marino, M. Molecular Mechanisms of Action of BPA. Dose-Response 2015, 13, 1559325815610582. [Google Scholar] [CrossRef] [Green Version]
- Martini, C.; Pallottini, V. Cholesterol: From feeding to gene regulation. Genes Nutr. 2007, 2, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.; Goldstein, J. Receptor-mediated control of cholesterol metabolism. Science 1976, 191, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Van De Sluis, B.; Wijers, M.; Herz, J. News on the molecular regulation and function of hepatic low-density lipoprotein receptor and LDLR-related protein 1. Curr. Opin. Lipidol. 2017, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Segatto, M.; Trapani, L.; Di Tunno, I.; Sticozzi, C.; Valacchi, G.; Hayek, J.; Pallottini, V. Cholesterol Metabolism Is Altered in Rett Syndrome: A Study on Plasma and Primary Cultured Fibroblasts Derived from Patients. PLoS ONE 2014, 9, e104834. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, K.; Lawn, R.M.; Wade, D.P. ABC1 Gene Expression and ApoA-I-Mediated Cholesterol Efflux Are Regulated by LXR. Biochem. Biophys. Res. Commun. 2000, 274, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Silver, D.L.; Costet, P.; Tall, A.R. Specific Binding of ApoA-I, Enhanced Cholesterol Efflux, and Altered Plasma Membrane Morphology in Cells Expressing ABC1. J. Biol. Chem. 2000, 275, 33053–33058. [Google Scholar] [CrossRef] [Green Version]
- Bloch, K. Sterol molecule: Structure, biosynthesis, and function. Steroids 1992, 57, 378–383. [Google Scholar] [CrossRef]
- Tian, L.; Li, W.; Yang, L.; Chang, N.; Fan, X.; Ji, X.; Xie, J.; Yang, L.; Li, L. Cannabinoid Receptor 1 Participates in Liver Inflammation by Promoting M1 Macrophage Polarization via RhoA/NF-κB p65 and ERK1/2 Pathways, Respectively, in Mouse Liver Fibrogenesis. Front. Immunol. 2017, 8, 1214. [Google Scholar] [CrossRef]
- Rinaudo, P.; Wang, E. Fetal Programming and Metabolic Syndrome. Annu. Rev. Physiol. 2012, 74, 107–130. [Google Scholar] [CrossRef] [Green Version]
- Heindel, J.J.; Skalla, L.A.; Joubert, B.; Dilworth, C.H.; Gray, K.A. Review of developmental origins of health and disease publications in environmental epidemiology. Reprod. Toxicol. 2017, 68, 34–48. [Google Scholar] [CrossRef]
- Benincasa, L.; Mandalà, M.; Paulesu, L.; Barberio, L.; Ietta, F. Prenatal Nutrition Containing Bisphenol A Affects Placenta Glucose Transfer: Evidence in Rats and Human Trophoblast. Nutrients 2020, 12, 1375. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Iwano, H.; Yanagisawa, R.; Koike, N.; Inoue, H.; Yokota, H. Placental Transfer of Conjugated Bisphenol A and Subsequent Reactivation in the Rat Fetus. Environ. Health Perspect. 2010, 118, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Ohlstein, J.F.; Strong, A.L.; A McLachlan, J.; Gimble, J.M.; E Burow, M.; A Bunnell, B. Bisphenol A enhances adipogenic differentiation of human adipose stromal/stem cells. J. Mol. Endocrinol. 2014, 53, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Lejonklou, M.H.; Dunder, L.; Bladin, E.; Pettersson, V.; Rönn, M.; Lind, L.; Waldén, T.B.; Lind, P.M. Effects of Low-Dose Developmental Bisphenol A Exposure on Metabolic Parameters and Gene Expression in Male and Female Fischer 344 Rat Offspring. Environ. Health Perspect. 2017, 125, 067018. [Google Scholar] [CrossRef]
- Dunder, L.; Lejonklou, M.H.; Lind, L.; Risérus, U.; Lind, P.M. Low-dose developmental bisphenol A exposure alters fatty acid metabolism in Fischer 344 rat offspring. Environ. Res. 2018, 166, 117–129. [Google Scholar] [CrossRef]
- Richter, C.A.; Birnbaum, L.S.; Farabollini, F.; Newbold, R.R.; Rubin, B.S.; Talsness, C.E.; Vandenbergh, J.G.; Walser-Kuntz, D.R.; Saal, F.S.V. In vivo effects of bisphenol A in laboratory rodent studies. Reprod. Toxicol. 2007, 24, 199–224. [Google Scholar] [CrossRef] [Green Version]
- Gabory, A.; Roseboom, T.J.; Moore, T.; Moore, L.G.; Junien, C. Placental contribution to the origins of sexual dimorphism in health and diseases: Sex chromosomes and epigenetics. Biol. Sex Differ. 2013, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babelova, A.; Burckhardt, B.C.; Salinas-Riester, G.; Pommerenke, C.; Burckhardt, G.; Henjakovic, M. Next generation sequencing of sex-specific genes in the livers of obese ZSF1 rats. Genomics 2015, 106, 204–213. [Google Scholar] [CrossRef]
- Link, J.C.; Reue, K. Genetic Basis for Sex Differences in Obesity and Lipid Metabolism. Annu. Rev. Nutr. 2017, 37, 225–245. [Google Scholar] [CrossRef]
- Somm, E.; Schwitzgebel, V.M.; Toulotte, A.; Cederroth, C.R.; Combescure, C.; Nef, S.; Aubert, M.L.; Hüppi, P. Perinatal Exposure to Bisphenol A Alters Early Adipogenesis in the Rat. Environ. Health Perspect. 2009, 117, 1549–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonini, C.; Segatto, M.; Pallottini, V. Impact of Sex and Age on the Mevalonate Pathway in the Brain: A Focus on Effects Induced by Maternal Exposure to Exogenous Compounds. Metabolites 2020, 10, 304. [Google Scholar] [CrossRef] [PubMed]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Subramaniam, S.; Dramane, G.; Khelifi, D.; Khan, N.A. ERK1 and ERK2 activation modulates diet-induced obesity in mice. Biochimie 2017, 137, 78–87. [Google Scholar] [CrossRef]
- Oba, D.; Inoue, S.-I.; Miyagawa-Tomita, S.; Nakashima, Y.; Niihori, T.; Yamaguchi, S.; Matsubara, Y.; Aoki, Y. Mice with an Oncogenic HRAS Mutation are Resistant to High-Fat Diet-Induced Obesity and Exhibit Impaired Hepatic Energy Homeostasis. EBioMedicine 2018, 27, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-C.; Jeong, W.-J.; Seo, S.H.; Choi, K.-Y. WDR76 mediates obesity and hepatic steatosis via HRas destabilization. Sci. Rep. 2019, 9, 19676. [Google Scholar] [CrossRef]
- Hall, D.J.; Cui, J.; Bates, M.E.; Stout, B.A.; Koenderman, L.; Coffer, P.J.; Bertics, P.J. Transduction of a dominant-negative H-Ras into human eosinophils attenuates extracellular signal–regulated kinase activation and interleukin-5–mediated cell viability. Blood 2001, 98, 2014–2021. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, T.; Okazaki, H.; Yudoh, K.; Matsuno, H.; Minota, S. Apoptosis of rheumatoid synovial cells by statins through the blocking of protein geranylgeranylation: A potential therapeutic approach to rheumatoid arthritis. Arthritis Rheum. 2006, 54, 579–586. [Google Scholar] [CrossRef]
- Jain, M.K.; Ridker, P.M. Anti-Inflammatory Effects of Statins: Clinical Evidence and Basic Mechanisms. Nat. Rev. Drug Discov. 2005, 4, 977–987. [Google Scholar] [CrossRef]
- Fracassi, A.; Marangoni, M.; Rosso, P.; Pallottini, V.; Fioramonti, M.; Siteni, S.; Segatto, M. Statins and the Brain: More than Lipid Lowering Agents? Curr. Neuropharmacol. 2018, 17, 59–83. [Google Scholar] [CrossRef]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NF B. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugazhenthi, S.; Zhang, Y.; Bouchard, R.; Mahaffey, G. Induction of an Inflammatory Loop by Interleukin-1β and Tumor Necrosis Factor-α Involves NF-kB and STAT-1 in Differentiated Human Neuroprogenitor Cells. PLoS ONE 2013, 8, e69585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elswefy, S.E.-S.; Abdallah, F.R.; Atteia, H.H.; Wahba, A.S.; Hasan, R.A. Inflammation, oxidative stress and apoptosis cascade implications in bisphenol A-induced liver fibrosis in male rats. Int. J. Exp. Pathol. 2016, 97, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.A.; Metz, L.; Yong, V.W. Review: Endocrine disrupting chemicals and immune responses: A focus on bisphenol-A and its potential mechanisms. Mol. Immunol. 2013, 53, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The fetal and infant origins of adult disease. BMJ 1990, 301, 1111. [Google Scholar] [CrossRef] [Green Version]
- McMillen, I.C.; Rattanatray, L.; Duffield, J.A.; Morrison, J.L.; MacLaughlin, S.M.; Gentili, S.; Muhlhäusler, B.S. The Early Origins of Later Obesity: Pathways and Mechanisms. Chem. Biol. Pteridines Folates 2009, 646, 71–81. [Google Scholar] [CrossRef]
- Malassiné, A.; Frendo, J.L.; Evain-Brion, D. A comparison of placental development and endocrine functions between the human and mouse model. Hum. Reprod. Update 2003, 9, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Free BPA (pg/mL) | TC (mg/dL) | TG (mg/dL) | HDL-C (mg/dL) | LDL-C (mg/dL) |
|---|---|---|---|---|---|
| CTR | 12.61 ± 0.92 | 88.00 ± 14.44 | 299.67 ± 104.86 | 61.33 ± 10.89 | 16.50 ± 9.42 |
| BPA | 13.98 ± 3.44 | 70.80 ± 17.41 | 366.20 ± 142.72 | 46.00 ± 12.21 | 10.00 ± 2.55 |
| Lipid Content | CTR F | BPA F |
|---|---|---|
| Total cholesterol (µg/mg tissue) | 2.78 ± 0.41 | 2.95 ± 1.01 |
| Free cholesterol (µg/mg tissue) | 2.51 ± 0.51 | 2.70 ± 0.75 |
| Triglycerides (nmol/mg tissue) | 2.03 ± 0.27 | 1.92 ± 0.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonini, C.; Segatto, M.; Bertoli, S.; Leone, A.; Mazzoli, A.; Cigliano, L.; Barberio, L.; Mandalà, M.; Pallottini, V. Prenatal Exposure to BPA: The Effects on Hepatic Lipid Metabolism in Male and Female Rat Fetuses. Nutrients 2021, 13, 1970. https://doi.org/10.3390/nu13061970
Tonini C, Segatto M, Bertoli S, Leone A, Mazzoli A, Cigliano L, Barberio L, Mandalà M, Pallottini V. Prenatal Exposure to BPA: The Effects on Hepatic Lipid Metabolism in Male and Female Rat Fetuses. Nutrients. 2021; 13(6):1970. https://doi.org/10.3390/nu13061970
Chicago/Turabian StyleTonini, Claudia, Marco Segatto, Simona Bertoli, Alessandro Leone, Arianna Mazzoli, Luisa Cigliano, Laura Barberio, Maurizio Mandalà, and Valentina Pallottini. 2021. "Prenatal Exposure to BPA: The Effects on Hepatic Lipid Metabolism in Male and Female Rat Fetuses" Nutrients 13, no. 6: 1970. https://doi.org/10.3390/nu13061970
APA StyleTonini, C., Segatto, M., Bertoli, S., Leone, A., Mazzoli, A., Cigliano, L., Barberio, L., Mandalà, M., & Pallottini, V. (2021). Prenatal Exposure to BPA: The Effects on Hepatic Lipid Metabolism in Male and Female Rat Fetuses. Nutrients, 13(6), 1970. https://doi.org/10.3390/nu13061970