
Vitamin D and Systems Biology


{kind=link}
Abstract
:1. Systems Biology and Biomedicine
1.1. The Opportunities of Applying Systems Biology Approaches in Biomedical Research
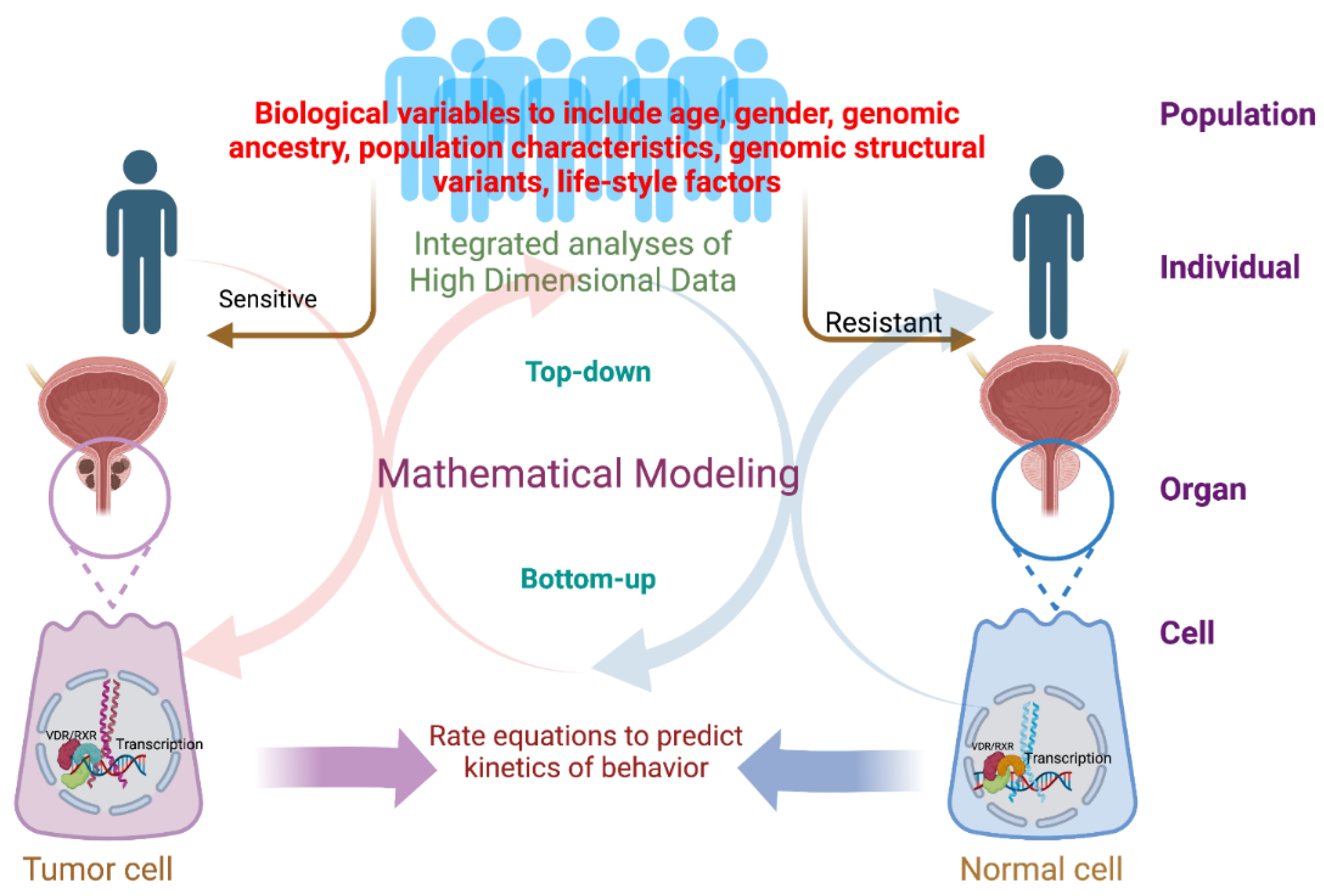
1.2. Systems Biology Builds upon an Asymptotic Recursion between Wet Lab and Dry Lab
2. The Opportunities and Challenges of Applying Systems Biology Approaches to Studying the Vitamin D Receptor
2.1. Top-Down Approaches Applied to VDR Biology
2.2. Bottom-Up Approaches Applied to VDR Biology
3. Prostate Cancer and Health Disparities; An Exemplar of the Opportunities Arising from Systems Biology Approaches in Biomedicine
The Potential Application of SB Approaches to Prostate Health and Disease
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rowley, J.D. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Lydon, N.B. Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J. Clin. Investig. 2000, 105, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Bixby, D.; Talpaz, M. Seeking the causes and solutions to imatinib-resistance in chronic myeloid leukemia. Leukemia 2011, 25, 7–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassman, M. Barriers to progress in systems biology. Nature 2005, 438, 1079. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.; Lischer, C.; Nenov, M.; Nikolov, S.; Lai, X.; Eberhardt, M. Mathematical Modelling in Biomedicine: A Primer for the Curious and the Skeptic. Int. J. Mol. Sci. 2021, 22, 547. [Google Scholar] [CrossRef] [PubMed]
- Haran, T.K.; Keren, L. From genes to modules, from cells to ecosystems. Mol. Syst. Biol. 2022, 18, e10726. [Google Scholar]
- Angione, C. Human Systems Biology and Metabolic Modelling: A Review-From Disease Metabolism to Precision Medicine. Biomed. Res. Int. 2019, 2019, 8304260. [Google Scholar] [CrossRef]
- Kuenzi, B.M.; Ideker, T. A census of pathway maps in cancer systems biology. Nat. Rev. Cancer 2020, 20, 233–246. [Google Scholar] [CrossRef]
- Wynn, M.L.; Consul, N.; Merajver, S.D.; Schnell, S. Logic-based models in systems biology: A predictive and parameter-free network analysis method. Integr. Biol. 2012, 4, 1323–1337. [Google Scholar] [CrossRef]
- Germain, R.N. Will Systems Biology Deliver Its Promise and Contribute to the Development of New or Improved Vaccines? What Really Constitutes the Study of “Systems Biology” and How Might Such an Approach Facilitate Vaccine Design. Cold Spring Harb. Perspect. Biol. 2018, 10, a033308. [Google Scholar] [CrossRef]
- Petrasek, D. Systems biology: The case for a systems science approach to diabetes. J. Diabetes Sci. Technol. 2008, 2, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Yamaguchi, S.; Yagita, K. Molecular machinery of the circadian clock in mammals. Cell Tissue Res. 2002, 309, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Chen, R.; Mo, Y.; Gu, S.; Ma, J.; Xu, W.; Lu, X.; He, H.; Jiang, F.; Fan, W.; et al. Quantitative control of noise in mammalian gene expression by dynamic histone regulation. Elife 2021, 10, e65654. [Google Scholar] [CrossRef] [PubMed]
- Thorne, J.L.; Campbell, M.J.; Turner, B.M. Transcription factors, chromatin and cancer. Int. J. Biochem. Cell Biol. 2009, 41, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Dunlop, T.W. An integrated biological approach to nuclear receptor signaling in physiological control and disease. Crit. Rev. Eukaryot. Gene Expr. 2006, 16, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Flores, M.; Glusman, G.; Brogaard, K.; Price, N.D.; Hood, L. P4 medicine: How systems medicine will transform the healthcare sector and society. Per. Med. 2013, 10, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Sobie, E.A.; Lee, Y.S.; Jenkins, S.L.; Iyengar, R. Systems biology—Biomedical modeling. Sci. Signal 2011, 4, tr2. [Google Scholar] [CrossRef] [Green Version]
- Westerhoff, H.V.; Palsson, B.O. The evolution of molecular biology into systems biology. Nat. Biotechnol. 2004, 22, 1249–1252. [Google Scholar] [CrossRef]
- Bruggeman, F.J.; Westerhoff, H.V. The nature of systems biology. Trends Microbiol. 2007, 15, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Snoep, J.L. The Silicon Cell initiative: Working towards a detailed kinetic description at the cellular level. Curr. Opin. Biotechnol. 2005, 16, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H. Daniel Whistler: English physician who published the first book on rickets in 1645. Br. J. Hosp. Med. 2019, 80, 51. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Campbell, M.J. Vitamin D receptor signaling mechanisms: Integrated actions of a well-defined transcription factor. Steroids 2013, 78, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Seuter, S.; Neme, A.; Carlberg, C. Characterization of genomic vitamin D receptor binding sites through chromatin looping and opening. PLoS ONE 2014, 9, e96184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, J.; Tabunoki, H. Molecular network of chromatin immunoprecipitation followed by deep sequencing-based vitamin D receptor target genes. Mult. Scler. 2013, 19, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Ramagopalan, S.V.; Heger, A.; Berlanga, A.J.; Maugeri, N.J.; Lincoln, M.R.; Burrell, A.; Handunnetthi, L.; Handel, A.E.; Disanto, G.; Orton, S.M.; et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: Associations with disease and evolution. Genome Res. 2010, 20, 1352–1360. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.B.; Goetsch, P.D.; Pike, J.W. VDR/RXR and TCF4/beta-catenin cistromes in colonic cells of colorectal tumor origin: Impact on c-FOS and c-MYC gene expression. Mol. Endocrinol. 2012, 26, 37–51. [Google Scholar] [CrossRef]
- Gonzalez-Duarte, R.J.; Cazares-Ordonez, V.; Diaz, L.; Ortiz, V.; Larrea, F.; Avila, E. The expression of RNA helicase DDX5 is transcriptionally upregulated by calcitriol through a vitamin D response element in the proximal promoter in SiHa cervical cells. Mol. Cell Biochem. 2015, 410, 65–73. [Google Scholar] [CrossRef]
- Wagner, M.; Rid, R.; Maier, C.J.; Maier, R.H.; Laimer, M.; Hintner, H.; Bauer, J.W.; Onder, K. DDX5 is a multifunctional co-activator of steroid hormone receptors. Mol. Cell Endocrinol. 2012, 361, 80–91. [Google Scholar] [CrossRef]
- MacDonald, P.N.; Dowd, D.R.; Zhang, C.; Gu, C. Emerging insights into the coactivator role of NCoA62/SKIP in Vitamin D-mediated transcription. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Foulds, C.E.; Panigrahi, A.K.; Coarfa, C.; Lanz, R.B.; O’Malley, B.W. Long Noncoding RNAs as Targets and Regulators of Nuclear Receptors. Curr. Top Microbiol. Immunol. 2016, 394, 143–176. [Google Scholar] [PubMed]
- Lanz, R.B.; Razani, B.; Goldberg, A.D.; O’Malley, B.W. Distinct RNA motifs are important for coactivation of steroid hormone receptors by steroid receptor RNA activator (SRA). Proc. Natl. Acad. Sci. USA 2002, 99, 16081–16086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Novershtern, N.; Subramanian, A.; Lawton, L.N.; Mak, R.H.; Haining, W.N.; McConkey, M.E.; Habib, N.; Yosef, N.; Chang, C.Y.; Shay, T.; et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 2011, 144, 296–309. [Google Scholar] [CrossRef] [Green Version]
- Novikova, S.; Tikhonova, O.; Kurbatov, L.; Farafonova, T.; Vakhrushev, I.; Lupatov, A.; Yarygin, K.; Zgoda, V. Omics Technologies to Decipher Regulatory Networks in Granulocytic Cell Differentiation. Biomolecules 2021, 11, 907. [Google Scholar] [CrossRef]
- Fuhrken, P.G.; Chen, C.; Apostolidis, P.A.; Wang, M.; Miller, W.M.; Papoutsakis, E.T. Gene Ontology-driven transcriptional analysis of CD34+ cell-initiated megakaryocytic cultures identifies new transcriptional regulators of megakaryopoiesis. Physiol. Genom. 2008, 33, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Yetgin, S.; Ozsoylu, S. Myeloid metaplasia in vitamin D deficiency rickets. Scand. J. Haematol. 1982, 28, 180–185. [Google Scholar] [CrossRef]
- Koeffler, H.P. Induction of differentiation of human acute myelogenous leukemia cells: Therapeutic implications. Blood 1983, 62, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Ji, Y.; Studzinski, G.P. Retinoblastoma protein and CCAAT/enhancer-binding protein beta are required for 1,25-dihydroxyvitamin D3-induced monocytic differentiation of HL60 cells. Cancer Res. 2004, 64, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Shiozaki, Y.; Miyazaki-Anzai, S.; Keenan, A.L.; Miyazaki, M. MEF2D-NR4A1-FAM134B2-mediated reticulophagy contributes to amino acid homeostasis. Autophagy 2022, 18, 1049–1061. [Google Scholar] [CrossRef]
- Marchwicka, A.; Marcinkowska, E. Regulation of Expression of CEBP Genes by Variably Expressed Vitamin D Receptor and Retinoic Acid Receptor alpha in Human Acute Myeloid Leukemia Cell Lines. Int. J. Mol. Sci. 2018, 19, 1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansure, J.J.; Nassim, R.; Chevalier, S.; Szymanski, K.; Rocha, J.; Aldousari, S.; Kassouf, W. A novel mechanism of PPAR gamma induction via EGFR signalling constitutes rational for combination therapy in bladder cancer. PLoS ONE 2013, 8, e55997. [Google Scholar] [CrossRef]
- Tang, Q.Q.; Otto, T.C.; Lane, M.D. CCAAT/enhancer-binding protein beta is required for mitotic clonal expansion during adipogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 850–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolotarenko, A.; Chekalin, E.; Mesentsev, A.; Kiseleva, L.; Gribanova, E.; Mehta, R.; Baranova, A.; Tatarinova, T.V.; Piruzian, E.S.; Bruskin, S. Integrated computational approach to the analysis of RNA-seq data reveals new transcriptional regulators of psoriasis. Exp. Mol. Med. 2016, 48, e268. [Google Scholar] [CrossRef] [Green Version]
- Greenhalgh, I.; Butler, A.R. Sanatoria revisited: Sunlight and health. J. R. Coll. Phys. Edinb. 2017, 47, 276–280. [Google Scholar] [CrossRef]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef]
- Mvubu, N.E.; Pillay, B.; Gamieldien, J.; Bishai, W.; Pillay, M. Canonical pathways, networks and transcriptional factor regulation by clinical strains of Mycobacterium tuberculosis in pulmonary alveolar epithelial cells. Tuberculosis 2016, 97, 73–85. [Google Scholar] [CrossRef]
- Sims, A.C.; Tilton, S.C.; Menachery, V.D.; Gralinski, L.E.; Schafer, A.; Matzke, M.M.; Webb-Robertson, B.J.; Chang, J.; Luna, M.L.; Long, C.E.; et al. Release of severe acute respiratory syndrome coronavirus nuclear import block enhances host transcription in human lung cells. J. Virol. 2013, 87, 3885–3902. [Google Scholar] [CrossRef] [Green Version]
- Chauss, D.; Freiwald, T.; McGregor, R.; Yan, B.; Wang, L.; Nova-Lamperti, E.; Kumar, D.; Zhang, Z.; Teague, H.; West, E.E.; et al. Autocrine vitamin D signaling switches off pro-inflammatory programs of TH1 cells. Nat. Immunol. 2022, 23, 62–74. [Google Scholar] [CrossRef]
- Adolphe, C.; Xue, A.; Fard, A.T.; Genovesi, L.A.; Yang, J.; Wainwright, B.J. Genetic and functional interaction network analysis reveals global enrichment of regulatory T cell genes influencing basal cell carcinoma susceptibility. Genome Med. 2021, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Thingholm, L.B.; Skieceviciene, J.; Rausch, P.; Kummen, M.; Hov, J.R.; Degenhardt, F.; Heinsen, F.A.; Ruhlemann, M.C.; Szymczak, S.; et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 2016, 48, 1396–1406. [Google Scholar] [CrossRef]
- Tavasolian, F.; Hosseini, A.Z.; Soudi, S.; Naderi, M.; Sahebkar, A. A Systems Biology Approach for miRNA-mRNA Expression Patterns Analysis in Rheumatoid Arthritis. Comb. Chem. High Throughput. Screen 2021, 24, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Montasser, M.E.; Aslibekyan, S.; Srinivasasainagendra, V.; Tiwari, H.K.; Patki, A.; Bagheri, M.; Kind, T.; Barupal, D.K.; Fan, S.; Perry, J.; et al. An Amish founder population reveals rare-population genetic determinants of the human lipidome. Commun. Biol. 2022, 5, 334. [Google Scholar] [CrossRef] [PubMed]
- Skinkyte-Juskiene, R.; Kogelman, L.J.A.; Kadarmideen, H.N. Transcription Factor Co-expression Networks of Adipose RNA-Seq Data Reveal Regulatory Mechanisms of Obesity. Curr. Genom. 2018, 19, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, M.H.; Luo, H.Q.; Chen, L.H.; Tang, L.; Ma, K.F.; Xiang, J.; Dong, L.P.; Zeng, J.; Li, G.Y.; Li, J.M. Impact of high fat diet on long non-coding RNAs and messenger RNAs expression in the aortas of ApoE(-/-) mice. Sci. Rep. 2016, 6, 34161. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.J. Bioinformatic approaches to interrogating vitamin D receptor signaling. Mol. Cell Endocrinol. 2017, 453, 3–13. [Google Scholar] [CrossRef]
- Vuckovic, D.; Bao, E.L.; Akbari, P.; Lareau, C.A.; Mousas, A.; Jiang, T.; Chen, M.H.; Raffield, L.M.; Tardaguila, M.; Huffman, J.E.; et al. The Polygenic and Monogenic Basis of Blood Traits and Diseases. Cell 2020, 182, 1214–1231.e1211. [Google Scholar] [CrossRef]
- Singh, P.K.; van den Berg, P.R.; Long, M.D.; Vreugdenhil, A.; Grieshober, L.; Ochs-Balcom, H.M.; Wang, J.; Delcambre, S.; Heikkinen, S.; Carlberg, C.; et al. Integration of VDR genome wide binding and GWAS genetic variation data reveals co-occurrence of VDR and NF-kappaB binding that is linked to immune phenotypes. BMC Genom. 2017, 18, 132. [Google Scholar] [CrossRef] [PubMed]
- Alleyne, D.; Witonsky, D.B.; Mapes, B.; Nakagome, S.; Sommars, M.; Hong, E.; Muckala, K.A.; Di Rienzo, A.; Kupfer, S.S. Colonic transcriptional response to 1alpha,25(OH)2 vitamin D3 in African- and European-Americans. J. Steroid Biochem. Mol. Biol. 2017, 168, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colak, Y.; Nordestgaard, B.G.; Afzal, S. Low vitamin D and risk of bacterial pneumonias: Mendelian randomisation studies in two population-based cohorts. Thorax 2021, 76, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Mazidi, M.; Davies, I.G.; Penson, P.; Rikkonen, T.; Isanejad, M. Lifetime serum concentration of 25-hydroxyvitamin D 25(OH) is associated with hand grip strengths: Insight from a Mendelian randomisation. Age Ageing 2022, 51, afac079. [Google Scholar] [CrossRef]
- Mokry, L.E.; Ross, S.; Ahmad, O.S.; Forgetta, V.; Smith, G.D.; Goltzman, D.; Leong, A.; Greenwood, C.M.; Thanassoulis, G.; Richards, J.B. Vitamin D and Risk of Multiple Sclerosis: A Mendelian Randomization Study. PLoS Med. 2015, 12, e1001866. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.S.; Luan, J.; Sofianopoulou, E.; Sharp, S.J.; Day, F.R.; Imamura, F.; Gundersen, T.E.; Lotta, L.A.; Sluijs, I.; Stewart, I.D.; et al. The association between circulating 25-hydroxyvitamin D metabolites and type 2 diabetes in European populations: A meta-analysis and Mendelian randomisation analysis. PLoS Med. 2020, 17, e1003394. [Google Scholar] [CrossRef]
- He, Y.; Timofeeva, M.; Farrington, S.M.; Vaughan-Shaw, P.; Svinti, V.; Walker, M.; Zgaga, L.; Meng, X.; Li, X.; Spiliopoulou, A.; et al. Exploring causality in the association between circulating 25-hydroxyvitamin D and colorectal cancer risk: A large Mendelian randomisation study. BMC Med. 2018, 16, 142. [Google Scholar] [CrossRef]
- Thompson, W.D.; Tyrrell, J.; Borges, M.C.; Beaumont, R.N.; Knight, B.A.; Wood, A.R.; Ring, S.M.; Hattersley, A.T.; Freathy, R.M.; Lawlor, D.A. Association of maternal circulating 25(OH)D and calcium with birth weight: A mendelian randomisation analysis. PLoS Med. 2019, 16, e1002828. [Google Scholar] [CrossRef] [Green Version]
- Tian, D.; Solodin, N.M.; Rajbhandari, P.; Bjorklund, K.; Alarid, E.T.; Kreeger, P.K. A kinetic model identifies phosphorylated estrogen receptor-alpha (ERalpha) as a critical regulator of ERalpha dynamics in breast cancer. FASEB J. 2015, 29, 2022–2031. [Google Scholar] [CrossRef] [Green Version]
- Cheong, R.; Hoffmann, A.; Levchenko, A. Understanding NF-kappaB signaling via mathematical modeling. Mol. Syst. Biol. 2008, 4, 192. [Google Scholar] [CrossRef]
- Bullock, M.E.; Moreno-Martinez, N.; Miller-Jensen, K. A transcriptional cycling model recapitulates chromatin-dependent features of noisy inducible transcription. PLoS Comput. Biol. 2022, 18, e1010152. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, A.; Lu, D.; Kalocsay, M.; Berberich, M.J.; Balbi, P.; Jambhekar, A.; Lahav, G. Time-series transcriptomics and proteomics reveal alternative modes to decode p53 oscillations. Mol. Syst. Biol. 2022, 18, e10588. [Google Scholar] [CrossRef] [PubMed]
- Ihekwaba, A.E.; Sedwards, S. Communicating oscillatory networks: Frequency domain analysis. BMC Syst. Biol. 2011, 5, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markevich, N.I.; Moehren, G.; Demin, O.V.; Kiyatkin, A.; Hoek, J.B.; Kholodenko, B.N. Signal processing at the Ras circuit: What shapes Ras activation patterns? Syst. Biol. 2004, 1, 104–113. [Google Scholar] [CrossRef]
- Stelniec-Klotz, I.; Legewie, S.; Tchernitsa, O.; Witzel, F.; Klinger, B.; Sers, C.; Herzel, H.; Bluthgen, N.; Schafer, R. Reverse engineering a hierarchical regulatory network downstream of oncogenic KRAS. Mol. Syst. Biol. 2012, 8, 601. [Google Scholar] [CrossRef]
- Relogio, A.; Thomas, P.; Medina-Perez, P.; Reischl, S.; Bervoets, S.; Gloc, E.; Riemer, P.; Mang-Fatehi, S.; Maier, B.; Schafer, R.; et al. Ras-mediated deregulation of the circadian clock in cancer. PLoS Genet. 2014, 10, e1004338. [Google Scholar] [CrossRef]
- Davies, A.E.; Pargett, M.; Siebert, S.; Gillies, T.E.; Choi, Y.; Tobin, S.J.; Ram, A.R.; Murthy, V.; Juliano, C.; Quon, G.; et al. Systems-Level Properties of EGFR-RAS-ERK Signaling Amplify Local Signals to Generate Dynamic Gene Expression Heterogeneity. Cell Syst. 2020, 11, 161–175.e165. [Google Scholar] [CrossRef]
- Patel, H.D.; Doshi, C.P.; Koehne, E.L.; Hart, S.; Van Kuiken, M.; Quek, M.L.; Flanigan, R.C.; Gupta, G.N. African American Men have Increased Risk of Prostate Cancer Detection Despite Similar Rates of Anterior Prostatic Lesions and PI-RADS Grade on Multiparametric Magnetic Resonance Imaging. Urology 2022, 163, 132–137. [Google Scholar] [CrossRef]
- Powell, I.J. Epidemiology and pathophysiology of prostate cancer in African-American men. J. Urol. 2007, 177, 444–449. [Google Scholar] [CrossRef]
- Moul, J.W.; Sesterhenn, I.A.; Connelly, R.R.; Douglas, T.; Srivastava, S.; Mostofi, F.K.; McLeod, D.G. Prostate-specific antigen values at the time of prostate cancer diagnosis in African-American men. JAMA 1995, 274, 1277–1281. [Google Scholar] [CrossRef]
- Sanchez-Ortiz, R.F.; Troncoso, P.; Babaian, R.J.; Lloreta, J.; Johnston, D.A.; Pettaway, C.A. African-American men with nonpalpable prostate cancer exhibit greater tumor volume than matched white men. Cancer 2006, 107, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Fiorica, P.N.; Schubert, R.; Morris, J.D.; Abdul Sami, M.; Wheeler, H.E. Multi-ethnic transcriptome-wide association study of prostate cancer. PLoS ONE 2020, 15, e0236209. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.J.; Van Den Eeden, S.K.; Sakoda, L.C.; Jorgenson, E.; Habel, L.A.; Graff, R.E.; Passarelli, M.N.; Cario, C.L.; Emami, N.C.; Chao, C.R.; et al. A large multiethnic genome-wide association study of prostate cancer identifies novel risk variants and substantial ethnic differences. Cancer Discov. 2015, 5, 878–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helfand, B.T.; Roehl, K.A.; Cooper, P.R.; McGuire, B.B.; Fitzgerald, L.M.; Cancel-Tassin, G.; Cornu, J.N.; Bauer, S.; Van Blarigan, E.L.; Chen, X.; et al. Associations of prostate cancer risk variants with disease aggressiveness: Results of the NCI-SPORE Genetics Working Group analysis of 18,343 cases. Hum. Genet. 2015, 134, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, J.O.; White, J.; Elhussin, I.; Duduyemi, B.M.; Karanam, B.; Yates, C.; Wang, H. Molecular and pathological subtypes related to prostate cancer disparities and disease outcomes in African American and European American patients. Front. Oncol. 2022, 12, 928357. [Google Scholar] [CrossRef]
- Awasthi, S.; Berglund, A.; Abraham-Miranda, J.; Rounbehler, R.J.; Kensler, K.; Serna, A.; Vidal, A.; You, S.; Freeman, M.R.; Davicioni, E.; et al. Comparative Genomics Reveals Distinct Immune-oncologic Pathways in African American Men with Prostate Cancer. Clin. Cancer Res. 2021, 27, 320–329. [Google Scholar] [CrossRef]
- Yuan, J.; Kensler, K.H.; Hu, Z.; Zhang, Y.; Zhang, T.; Jiang, J.; Xu, M.; Pan, Y.; Long, M.; Montone, K.T.; et al. Integrative comparison of the genomic and transcriptomic landscape between prostate cancer patients of predominantly African or European genetic ancestry. PLoS Genet. 2020, 16, e1008641. [Google Scholar] [CrossRef] [Green Version]
- Hardiman, G.; Savage, S.J.; Hazard, E.S.; Wilson, R.C.; Courtney, S.M.; Smith, M.T.; Hollis, B.W.; Halbert, C.H.; Gattoni-Celli, S. Systems analysis of the prostate transcriptome in African-American men compared with European-American men. Pharmacogenomics 2016, 17, 1129–1143. [Google Scholar] [CrossRef]
- Yamoah, K.; Asamoah, F.A.; Abrahams, A.O.D.; Awasthi, S.; Mensah, J.E.; Dhillon, J.; Mahal, B.A.; Gueye, S.M.; Jalloh, M.; Farahani, S.J.; et al. Prostate tumors of native men from West Africa show biologically distinct pathways-A comparative genomic study. Prostate 2021, 81, 1402–1410. [Google Scholar] [CrossRef]
- Koga, Y.; Song, H.; Chalmers, Z.R.; Newberg, J.; Kim, E.; Carrot-Zhang, J.; Piou, D.; Polak, P.; Abdulkadir, S.A.; Ziv, E.; et al. Genomic Profiling of Prostate Cancers from Men with African and European Ancestry. Clin. Cancer Res. 2020, 26, 4651–4660. [Google Scholar] [CrossRef]
- Rayford, W.; Beksac, A.T.; Alger, J.; Alshalalfa, M.; Ahmed, M.; Khan, I.; Falagario, U.G.; Liu, Y.; Davicioni, E.; Spratt, D.E.; et al. Comparative analysis of 1152 African-American and European-American men with prostate cancer identifies distinct genomic and immunological differences. Commun. Biol. 2021, 4, 670. [Google Scholar] [CrossRef]
- Liu, W.; Zheng, S.L.; Na, R.; Wei, L.; Sun, J.; Gallagher, J.; Wei, J.; Resurreccion, W.K.; Ernst, S.; Sfanos, K.S.; et al. Distinct Genomic Alterations in Prostate Tumors Derived from African American Men. Mol. Cancer Res. 2020, 18, 1815–1824. [Google Scholar] [CrossRef]
- Magi-Galluzzi, C.; Tsusuki, T.; Elson, P.; Simmerman, K.; LaFargue, C.; Esgueva, R.; Klein, E.; Rubin, M.A.; Zhou, M. TMPRSS2-ERG gene fusion prevalence and class are significantly different in prostate cancer of Caucasian, African-American and Japanese patients. Prostate 2011, 71, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Kerkvliet, C.P.; Truong, T.H.; Ostrander, J.H.; Lange, C.A. Stress sensing within the breast tumor microenvironment: How glucocorticoid receptors live in the moment. Essays Biochem. 2021, 65, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Rittschof, C.C.; Greenlee, A.J.; Turi, K.N.; Rodriguez-Zas, S.L.; Robinson, G.E.; Cole, S.W.; Mendenhall, R. Transcriptomic analyses of black women in neighborhoods with high levels of violence. Psychoneuroendocrinology 2021, 127, 105174. [Google Scholar] [CrossRef]
- Woods-Burnham, L.; Stiel, L.; Martinez, S.R.; Sanchez-Hernandez, E.S.; Ruckle, H.C.; Almaguel, F.G.; Stern, M.C.; Roberts, L.R.; Williams, D.R.; Montgomery, S.; et al. Psychosocial Stress, Glucocorticoid Signaling, and Prostate Cancer Health Disparities in African American Men. Cancer Health Disparities 2020, 4, e1–e30. [Google Scholar]
- Woods-Burnham, L.; Cajigas-Du Ross, C.K.; Love, A.; Basu, A.; Sanchez-Hernandez, E.S.; Martinez, S.R.; Ortiz-Hernandez, G.L.; Stiel, L.; Duran, A.M.; Wilson, C.; et al. Glucocorticoids Induce Stress Oncoproteins Associated with Therapy-Resistance in African American and European American Prostate Cancer Cells. Sci. Rep. 2018, 8, 15063. [Google Scholar] [CrossRef] [Green Version]
- Hochberg, Z.; Templeton, A.R. Evolutionary perspective in skin color, vitamin D and its receptor. Hormones 2010, 9, 307–311. [Google Scholar] [CrossRef] [Green Version]
- Jablonski, N.G. The evolution of human skin colouration and its relevance to health in the modern world. J. R. Coll. Phys. Edinb. 2012, 42, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Hong, C.C.; Bandera, E.V.; Zhu, Q.; Liu, S.; Cheng, T.D.; Zirpoli, G.; Haddad, S.A.; Lunetta, K.L.; Ruiz-Narvaez, E.A.; et al. Demographic, lifestyle, and genetic determinants of circulating concentrations of 25-hydroxyvitamin D and vitamin D-binding protein in African American and European American women. Am. J. Clin. Nutr. 2017, 105, 1362–1371. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Haddad, S.A.; Hu, Q.; Liu, S.; Lunetta, K.L.; Ruiz-Narvaez, E.A.; Hong, C.C.; Zhu, Q.; Sucheston-Campbell, L.; Cheng, T.Y.; et al. Genetic variations in vitamin D-related pathways and breast cancer risk in African American women in the AMBER consortium. Int. J. Cancer 2016, 138, 2118–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, D.K.; Wu, Y.; Sarkissyan, M.; Sarkissyan, S.; Chen, Z.; Shang, X.; Ong, M.; Heber, D.; Koeffler, H.P.; Vadgama, J.V. Vitamin D receptor gene polymorphisms and prognosis of breast cancer among African-American and Hispanic women. PLoS ONE 2013, 8, e57967. [Google Scholar] [CrossRef] [PubMed]
- Neuhouser, M.L.; Bernstein, L.; Hollis, B.W.; Xiao, L.; Ambs, A.; Baumgartner, K.; Baumgartner, R.; McTiernan, A.; Ballard-Barbash, R. Serum vitamin D and breast density in breast cancer survivors. Cancer Epidemiol. Biomark. Prev. 2010, 19, 412–417. [Google Scholar] [CrossRef] [Green Version]
- John, E.M.; Schwartz, G.G.; Koo, J.; Wang, W.; Ingles, S.A. Sun exposure, vitamin D receptor gene polymorphisms, and breast cancer risk in a multiethnic population. Am. J. Epidemiol. 2007, 166, 1409–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layne, T.M.; Weinstein, S.J.; Graubard, B.I.; Ma, X.; Mayne, S.T.; Albanes, D. Serum 25-hydroxyvitamin D, vitamin D binding protein, and prostate cancer risk in black men. Cancer 2017, 123, 2698–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, S.M.; Batai, K.; Ahaghotu, C.; Agurs-Collins, T.; Kittles, R.A. Association between Serum 25-Hydroxy-Vitamin D and Aggressive Prostate Cancer in African American Men. Nutrients 2016, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Batai, K.; Murphy, A.B.; Nonn, L.; Kittles, R.A. Vitamin D and Immune Response: Implications for Prostate Cancer in African Americans. Front. Immunol. 2016, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.B.; Nyame, Y.; Martin, I.K.; Catalona, W.J.; Hollowell, C.M.; Nadler, R.B.; Kozlowski, J.M.; Perry, K.T.; Kajdacsy-Balla, A.; Kittles, R. Vitamin D deficiency predicts prostate biopsy outcomes. Clin. Cancer Res. 2014, 20, 2289–2299. [Google Scholar] [CrossRef] [Green Version]
- Richards, Z.; Batai, K.; Farhat, R.; Shah, E.; Makowski, A.; Gann, P.H.; Kittles, R.; Nonn, L. Prostatic compensation of the vitamin D axis in African American men. JCI Insight 2017, 2, e91054. [Google Scholar] [CrossRef] [Green Version]
- Kidd, L.C.; Paltoo, D.N.; Wang, S.; Chen, W.; Akereyeni, F.; Isaacs, W.; Ahaghotu, C.; Kittles, R. Sequence variation within the 5′ regulatory regions of the vitamin D binding protein and receptor genes and prostate cancer risk. Prostate 2005, 64, 272–282. [Google Scholar] [CrossRef]
- Murphy, A.B.; Kelley, B.; Nyame, Y.A.; Martin, I.K.; Smith, D.J.; Castaneda, L.; Zagaja, G.J.; Hollowell, C.M.; Kittles, R.A. Predictors of serum vitamin D levels in African American and European American men in Chicago. Am. J. Mens. Health 2012, 6, 420–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paller, C.J.; Kanaan, Y.M.; Beyene, D.A.; Naab, T.J.; Copeland, R.L.; Tsai, H.L.; Kanarek, N.F.; Hudson, T.S. Risk of prostate cancer in African-American men: Evidence of mixed effects of dietary quercetin by serum vitamin D status. Prostate 2015, 75, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Batai, K.; Kittles, R.A. Can vitamin D supplementation reduce prostate cancer disparities? Pharmacogenomics 2016, 17, 1117–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taksler, G.B.; Cutler, D.M.; Giovannucci, E.; Smith, M.R.; Keating, N.L. Ultraviolet index and racial differences in prostate cancer incidence and mortality. Cancer 2013, 119, 3195–3203. [Google Scholar] [CrossRef]
- Steck, S.E.; Arab, L.; Zhang, H.; Bensen, J.T.; Fontham, E.T.; Johnson, C.S.; Mohler, J.L.; Smith, G.J.; Su, J.L.; Trump, D.L.; et al. Association between Plasma 25-Hydroxyvitamin D, Ancestry and Aggressive Prostate Cancer among African Americans and European Americans in PCaP. PLoS ONE 2015, 10, e0125151. [Google Scholar] [CrossRef] [Green Version]
- King, L.; Dear, K.; Harrison, S.L.; van der Mei, I.; Brodie, A.M.; Kimlin, M.G.; Lucas, R.M. Investigating the patterns and determinants of seasonal variation in vitamin D status in Australian adults: The Seasonal D Cohort Study. BMC Public Health 2016, 16, 892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diffey, B.L. Modelling the seasonal variation of vitamin D due to sun exposure. Br. J. Dermatol. 2010, 162, 1342–1348. [Google Scholar] [CrossRef]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Albert, C.M.; Gordon, D.; Copeland, T.; et al. Marine n-3 Fatty Acids and Prevention of Cardiovascular Disease and Cancer. N. Engl. J. Med. 2019, 380, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Bassuk, S.S.; Chandler, P.D.; Buring, J.E.; Manson, J.E.; Group, V.R. The VITamin D and OmegA-3 TriaL (VITAL): Do Results Differ by Sex or Race/Ethnicity? Am. J. Lifestyle Med. 2021, 15, 372–391. [Google Scholar] [CrossRef]
- Chandler, P.D.; Chen, W.Y.; Ajala, O.N.; Hazra, A.; Cook, N.; Bubes, V.; Lee, I.M.; Giovannucci, E.L.; Willett, W.; Buring, J.E.; et al. Effect of Vitamin D3 Supplements on Development of Advanced Cancer: A Secondary Analysis of the VITAL Randomized Clinical Trial. JAMA Netw. Open 2020, 3, e2025850. [Google Scholar] [CrossRef]
- Marshall, D.T.; Savage, S.J.; Garrett-Mayer, E.; Keane, T.E.; Hollis, B.W.; Horst, R.L.; Ambrose, L.H.; Kindy, M.S.; Gattoni-Celli, S. Vitamin D3 supplementation at 4000 international units per day for one year results in a decrease of positive cores at repeat biopsy in subjects with low-risk prostate cancer under active surveillance. J. Clin. Endocrinol. Metab. 2012, 97, 2315–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddappa, M.; Hussain, S.; Wani, S.A.; Tang, H.; Gray, J.S.; Jafari, H.; Wu, H.; Long, M.D.; Elhussin, I.; Karanam, B.; et al. Vitamin D receptor cistrome-transcriptome analyses establishes quantitatively distinct receptor genomic interactions in African American prostate cancer regulated by BAZ1A. bioRxiv 2022. [Google Scholar] [CrossRef]
- Singh, A.K.; Prakash, S.; Garg, R.K.; Jain, P.; Kumar, R.; Jain, A. Polymorphisms in vitamin D receptor, toll-like receptor 2 and Toll-Like receptor 4 genes links with Dengue susceptibility. Bioinformation 2021, 17, 506–513. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhou, M.; Li, Y.C. Vitamin D/Vitamin D Receptor Signaling Is Required for Normal Development and Function of Group 3 Innate Lymphoid Cells in the Gut. iScience 2019, 17, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konya, V.; Czarnewski, P.; Forkel, M.; Rao, A.; Kokkinou, E.; Villablanca, E.J.; Almer, S.; Lindforss, U.; Friberg, D.; Hoog, C.; et al. Vitamin D downregulates the IL-23 receptor pathway in human mucosal group 3 innate lymphoid cells. J. Allergy Clin. Immunol. 2018, 141, 279–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gombart, A.F.; Borregaard, N.; Koeffler, H.P. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J. 2005, 19, 1067–1077. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.L.; Lai, S.H.; Tsai, M.H.; Hua, M.C.; Yeh, K.W.; Su, K.W.; Chiang, C.H.; Huang, S.Y.; Kao, C.C.; Yao, T.C.; et al. Maternal Vitamin D Level Is Associated with Viral Toll-Like Receptor Triggered IL-10 Response but Not the Risk of Infectious Diseases in Infancy. Mediat. Inflamm. 2016, 2016, 8175898. [Google Scholar] [CrossRef] [Green Version]
- Ojaimi, S.; Skinner, N.A.; Strauss, B.J.; Sundararajan, V.; Woolley, I.; Visvanathan, K. Vitamin D deficiency impacts on expression of toll-like receptor-2 and cytokine profile: A pilot study. J. Transl. Med. 2013, 11, 176. [Google Scholar] [CrossRef] [Green Version]
- Darko, S.N.; Yar, D.D.; Owusu-Dabo, E.; Awuah, A.A.; Dapaah, W.; Addofoh, N.; Salifu, S.P.; Awua-Boateng, N.Y.; Adomako-Boateng, F. Variations in levels of IL-6 and TNF-alpha in type 2 diabetes mellitus between rural and urban Ashanti Region of Ghana. BMC Endocr. Disord. 2015, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Mensah, G.I.; Addo, K.K.; Tetteh, J.A.; Sowah, S.; Loescher, T.; Geldmacher, C.; Jackson-Sillah, D. Cytokine response to selected MTB antigens in Ghanaian TB patients, before and at 2 weeks of anti-TB therapy is characterized by high expression of IFN-gamma and Granzyme B and inter- individual variation. BMC Infect. Dis. 2014, 14, 495. [Google Scholar] [CrossRef] [Green Version]
- Minas, T.Z.; Candia, J.; Dorsey, T.H.; Baker, F.; Tang, W.; Kiely, M.; Smith, C.J.; Zhang, A.L.; Jordan, S.V.; Obadi, O.M.; et al. Serum proteomics links suppression of tumor immunity to ancestry and lethal prostate cancer. Nat. Commun. 2022, 13, 1759. [Google Scholar] [CrossRef]
- Chen, F.; Darst, B.F.; Madduri, R.K.; Rodriguez, A.A.; Sheng, X.; Rentsch, C.T.; Andrews, C.; Tang, W.; Kibel, A.S.; Plym, A.; et al. Validation of a multi-ancestry polygenic risk score and age-specific risks of prostate cancer: A meta-analysis within diverse populations. Elife 2022, 11, e78304. [Google Scholar] [CrossRef] [PubMed]
- Conti, D.V.; Darst, B.F.; Moss, L.C.; Saunders, E.J.; Sheng, X.; Chou, A.; Schumacher, F.R.; Olama, A.A.A.; Benlloch, S.; Dadaev, T.; et al. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat. Genet. 2021, 53, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Berchuck, J.E.; Adib, E.; Abou Alaiwi, S.; Dash, A.K.; Shin, J.N.; Lowder, D.; McColl, C.; Castro, P.; Carelli, R.; Benedetti, E.; et al. The prostate cancer androgen receptor cistrome in African American men associates with upregulation of lipid metabolism and immune response. Cancer Res. 2022, 82, 2848–2859. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef] [Green Version]
- Long, M.D.; Jacobi, J.J.; Singh, P.K.; Llimos, G.; Wani, S.A.; Rowsam, A.M.; Rosario, S.R.; Hoogstraat, M.; Linder, S.; Kirk, J.; et al. Reduced NCOR2 expression accelerates androgen deprivation therapy failure in prostate cancer. Cell Rep. 2021, 37, 110109. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Qiu, X.; Zhu, Y.; Takeda, D.Y.; Pan, W.; Baca, S.C.; Gusev, A.; Korthauer, K.D.; Severson, T.M.; Ha, G.; et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat. Genet. 2020, 52, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Long, M.D.; van den Berg, P.R.; Russell, J.L.; Singh, P.K.; Battaglia, S.; Campbell, M.J. Integrative genomic analysis in K562 chronic myelogenous leukemia cells reveals that proximal NCOR1 binding positively regulates genes that govern erythroid differentiation and Imatinib sensitivity. Nucleic Acids Res. 2015, 43, 7330–7348. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Kitanovski, S.; Hoffmann, D. intePareto: An R package for integrative analyses of RNA-Seq and ChIP-Seq data. BMC Genom. 2020, 21, 802. [Google Scholar] [CrossRef]
- Liu, J.; Lei, B.; Yu, X.; Li, Y.; Deng, Y.; Yang, G.; Li, Z.; Liu, T.; Ye, L. Combining Immune-Related Genes For Delineating the Extracellular Matrix and Predicting Hormone Therapy and Neoadjuvant Chemotherapy Benefits In Breast Cancer. Front. Immunol. 2022, 13, 888339. [Google Scholar] [CrossRef] [PubMed]
- Frost, H.R.; Amos, C.I. Gene set selection via LASSO penalized regression (SLPR). Nucleic Acids Res. 2017, 45, e114. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, S.; Yates, C.; Campbell, M.J. Vitamin D and Systems Biology. Nutrients 2022, 14, 5197. https://doi.org/10.3390/nu14245197
Hussain S, Yates C, Campbell MJ. Vitamin D and Systems Biology. Nutrients. 2022; 14(24):5197. https://doi.org/10.3390/nu14245197
Chicago/Turabian StyleHussain, Shahid, Clayton Yates, and Moray J. Campbell. 2022. "Vitamin D and Systems Biology" Nutrients 14, no. 24: 5197. https://doi.org/10.3390/nu14245197
APA StyleHussain, S., Yates, C., & Campbell, M. J. (2022). Vitamin D and Systems Biology. Nutrients, 14(24), 5197. https://doi.org/10.3390/nu14245197