

The Involvement of Intestinal Tryptophan Metabolism in Inflammatory Bowel Disease Identified by a Meta-Analysis of the Transcriptome and a Systematic Review of the Metabolome

Abstract
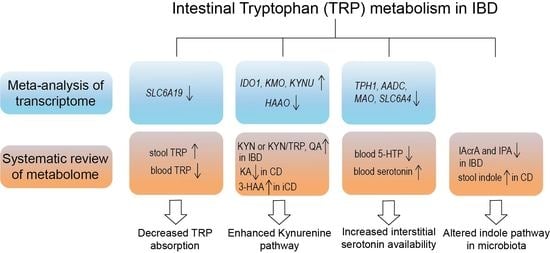
:
1. Introduction
2. Materials and Methods
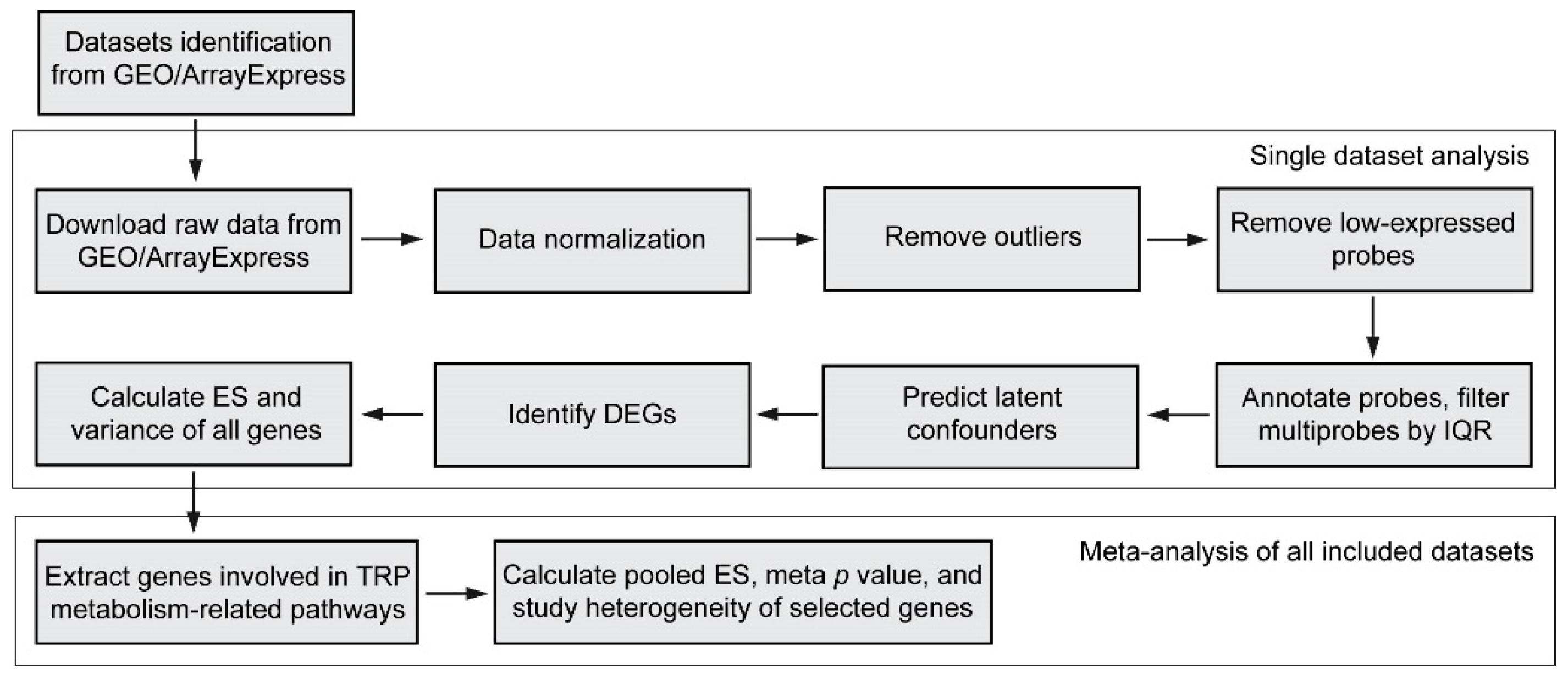
2.1. Meta-Analysis of the Transcriptome
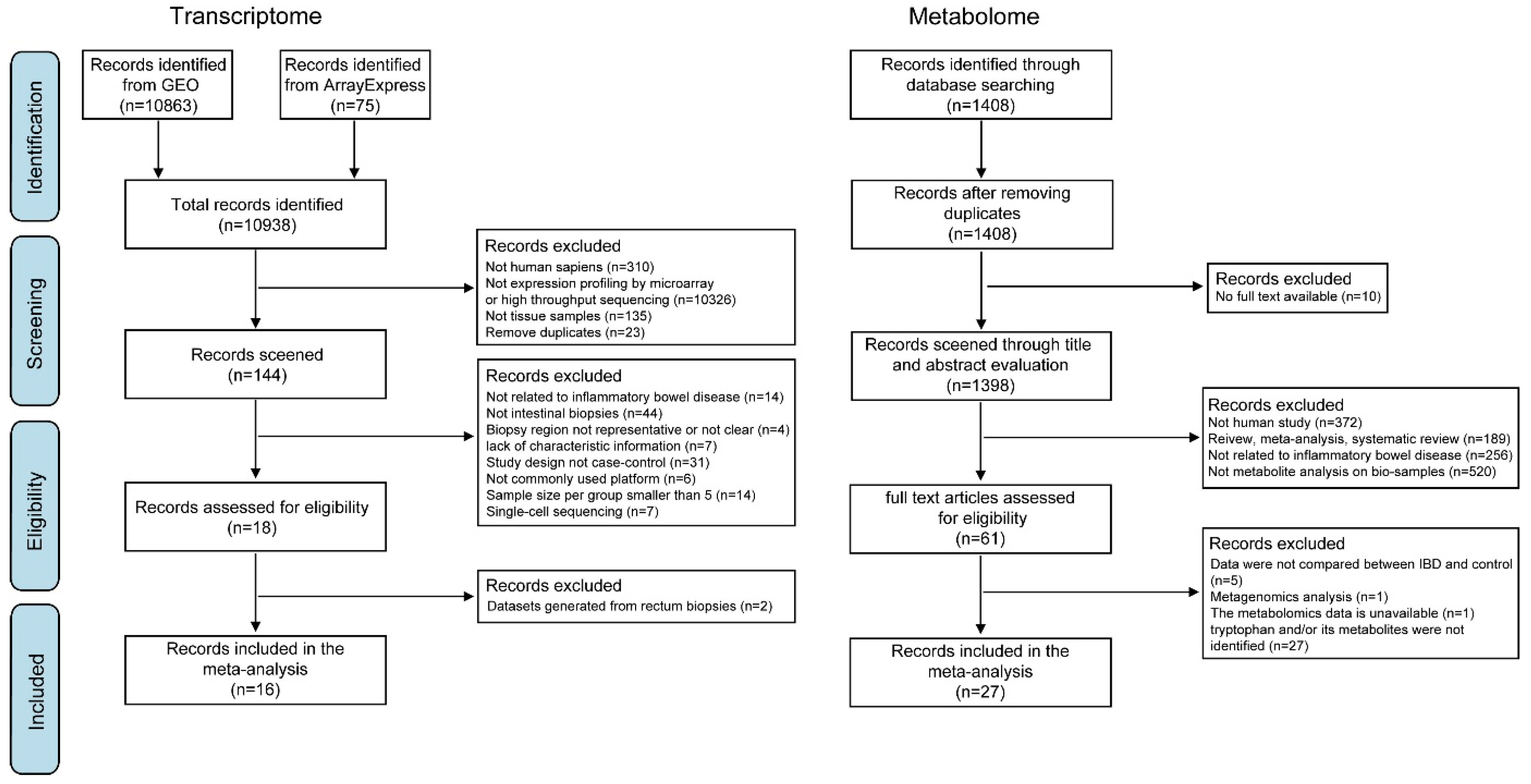
2.1.1. Systematic Review of the Transcriptome
2.1.2. Inclusion and Exclusion Criteria
2.1.3. Processing of a Single Transcriptomics Dataset
2.1.4. Gene Selection
2.1.5. Meta-Analysis
2.2. Systematic Review of the Metabolome
3. Results and Discussion
3.1. Systematic Review of the Transcriptome and Metabolome
3.2. Meta-Analysis of Gene Expressions and Summary of Metabolites Involved in Intestinal TRP Metabolism
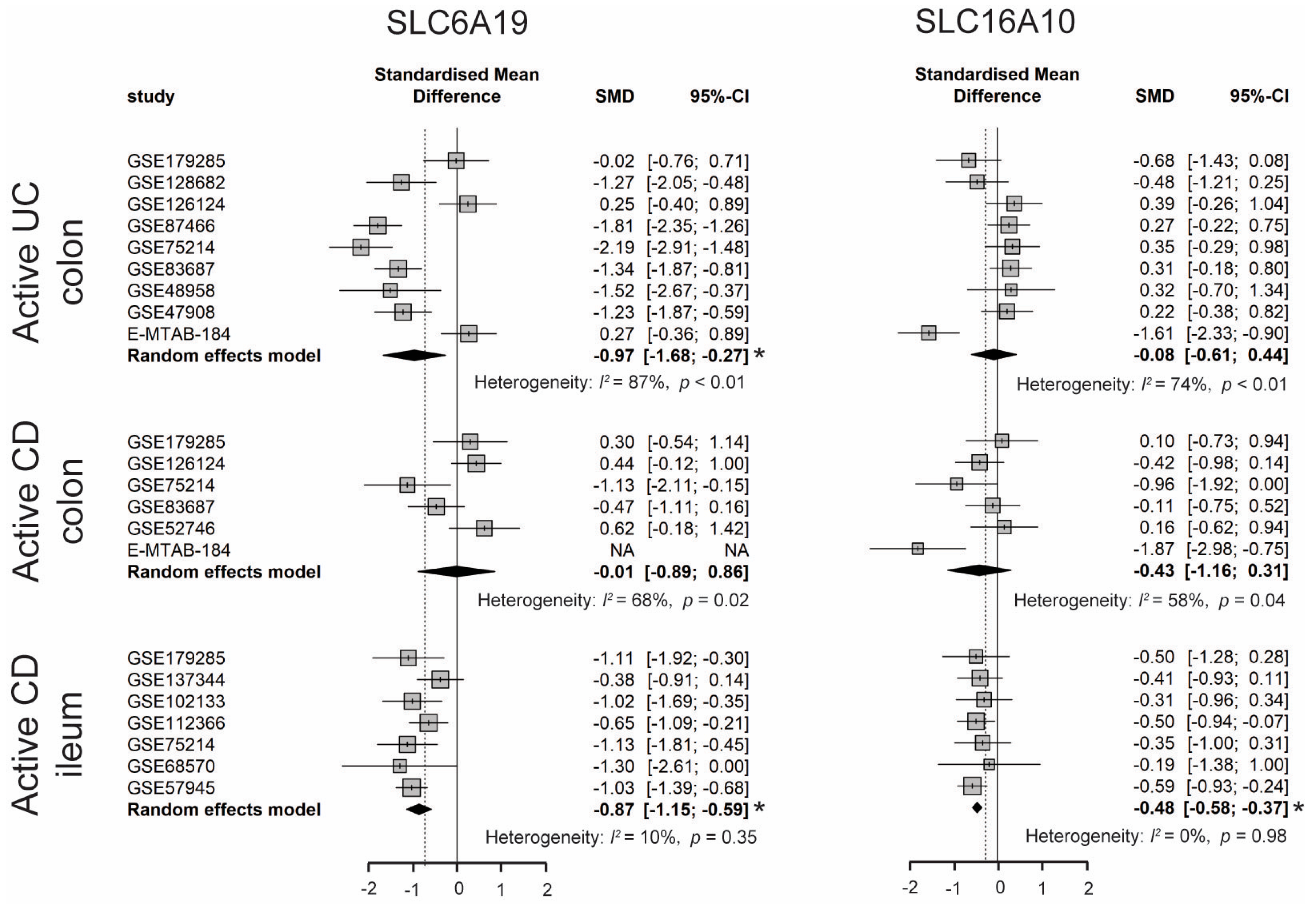
3.2.1. Decreased TRP Absorption in IBD Patients
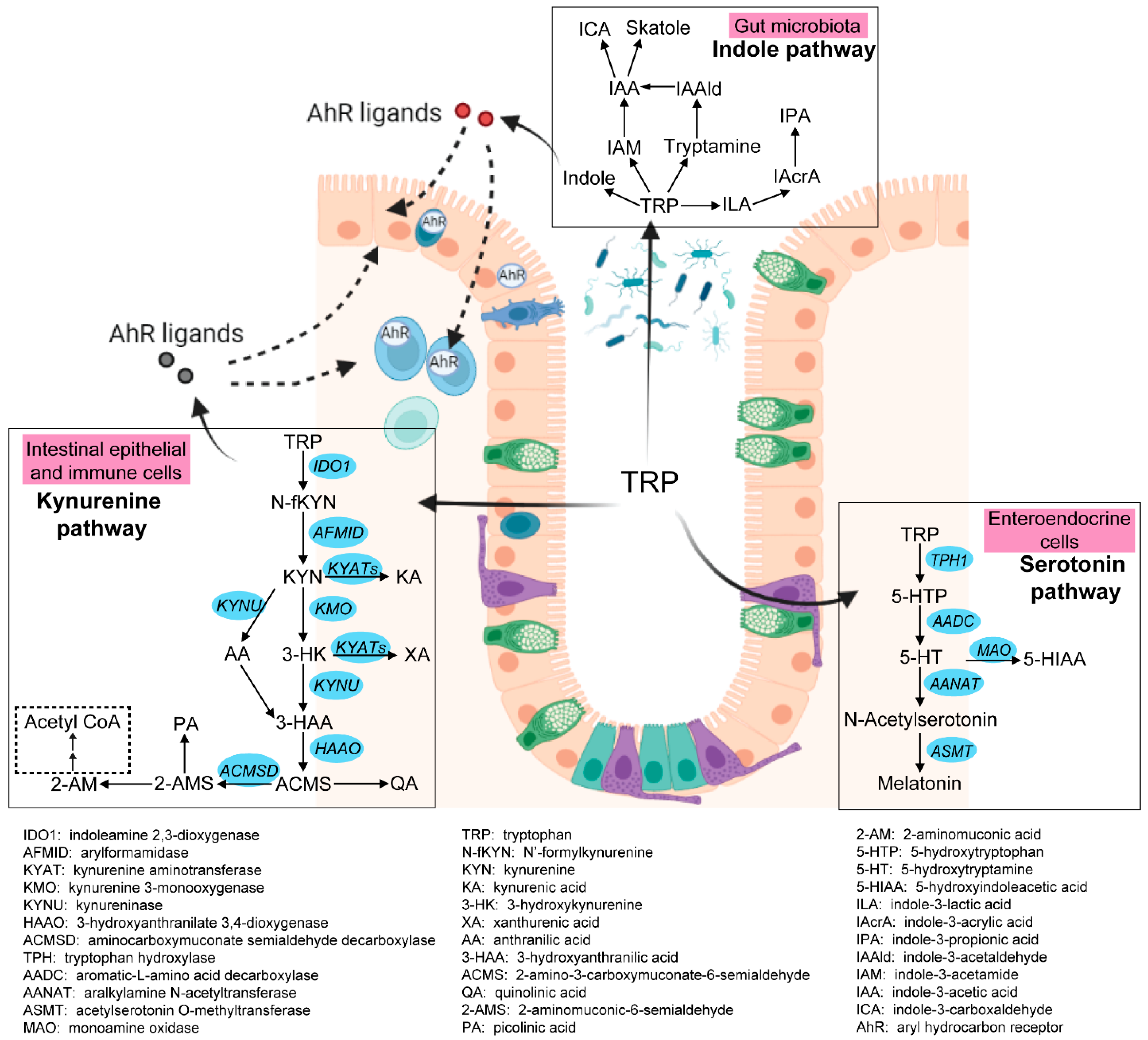
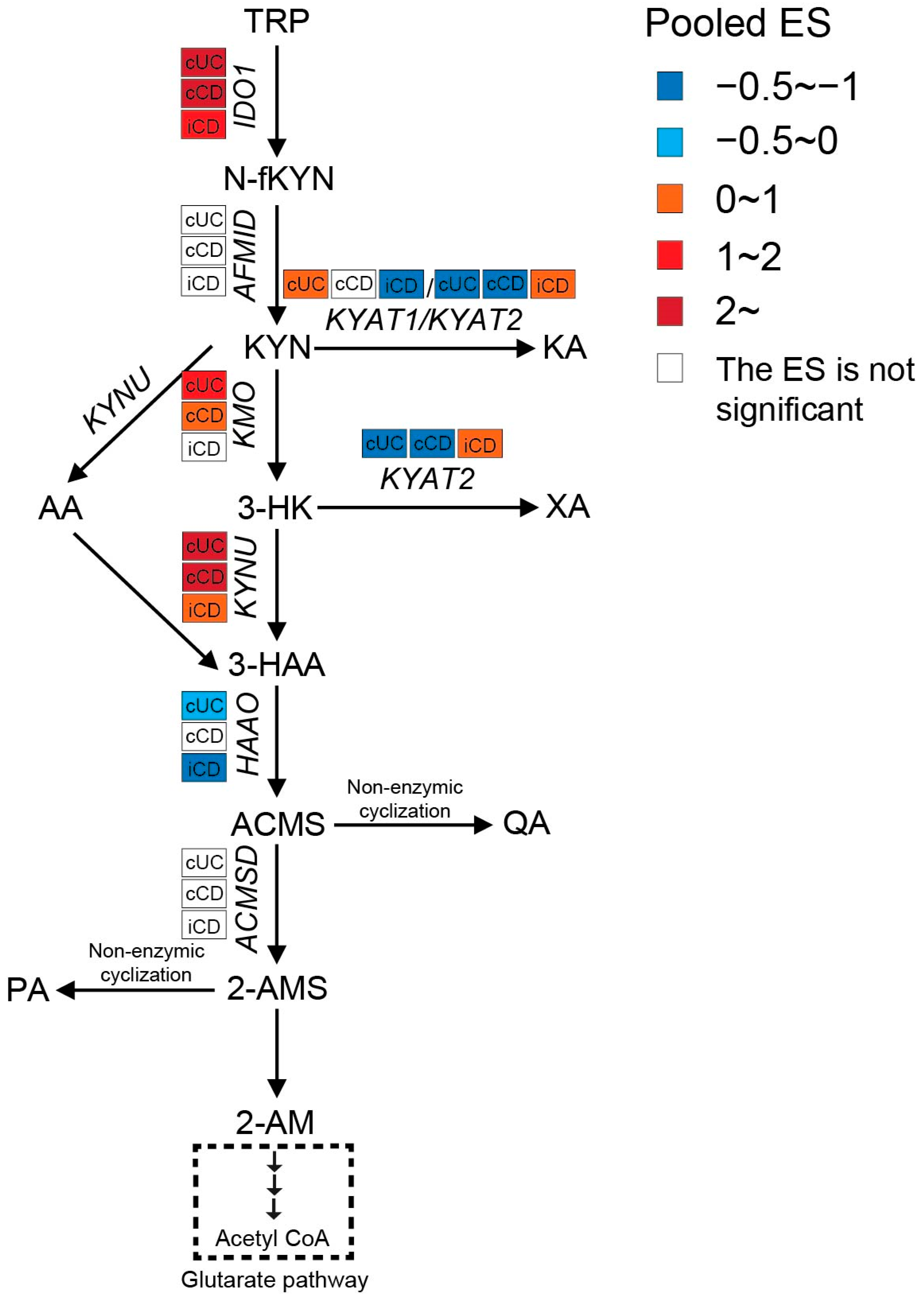
3.2.2. Enhanced Kynurenine Pathway (KP) in IBD Patients
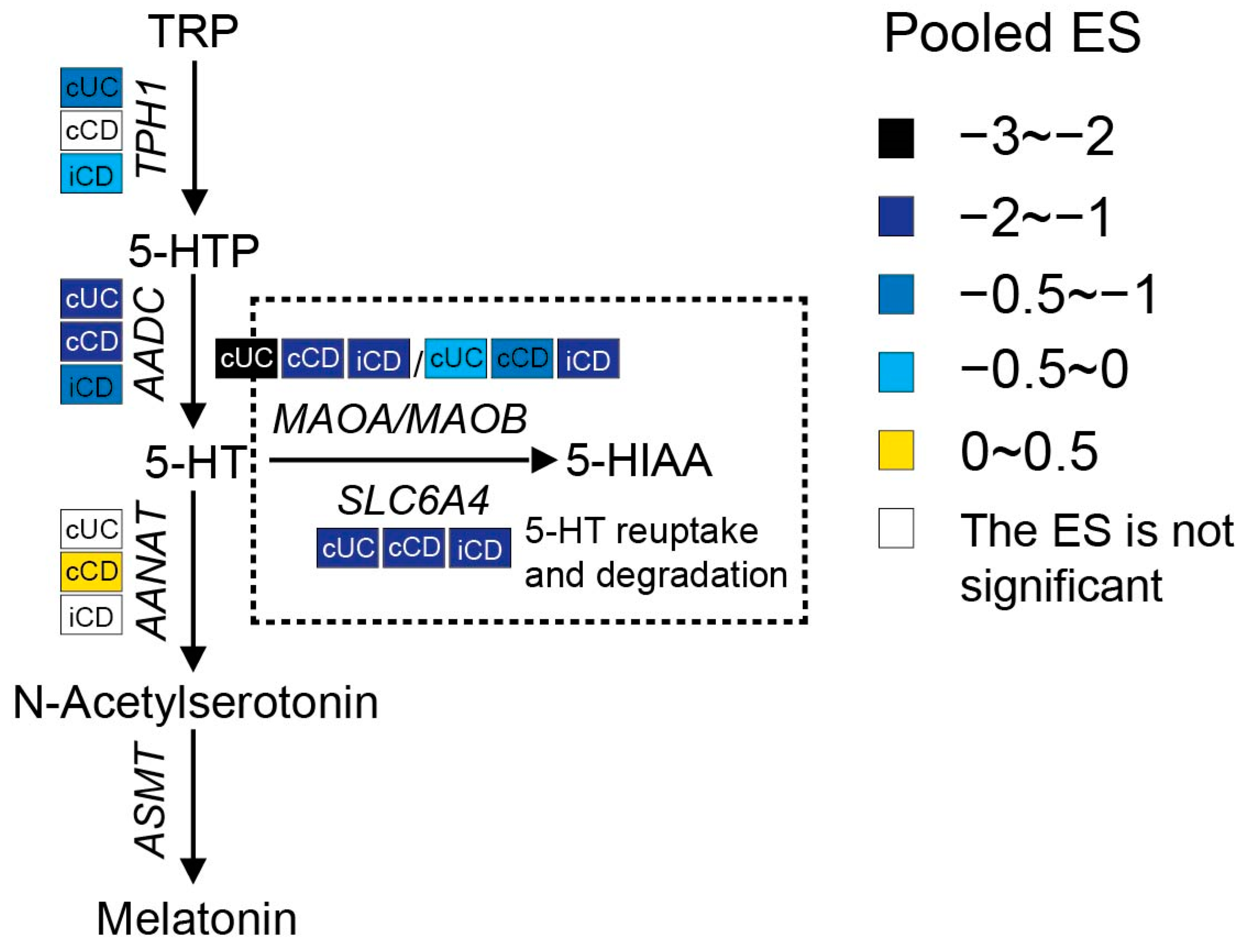
3.2.3. Increased Interstitial Serotonin Availability in IBD Patients
3.2.4. Altered Indole Pathway in IBD Patients
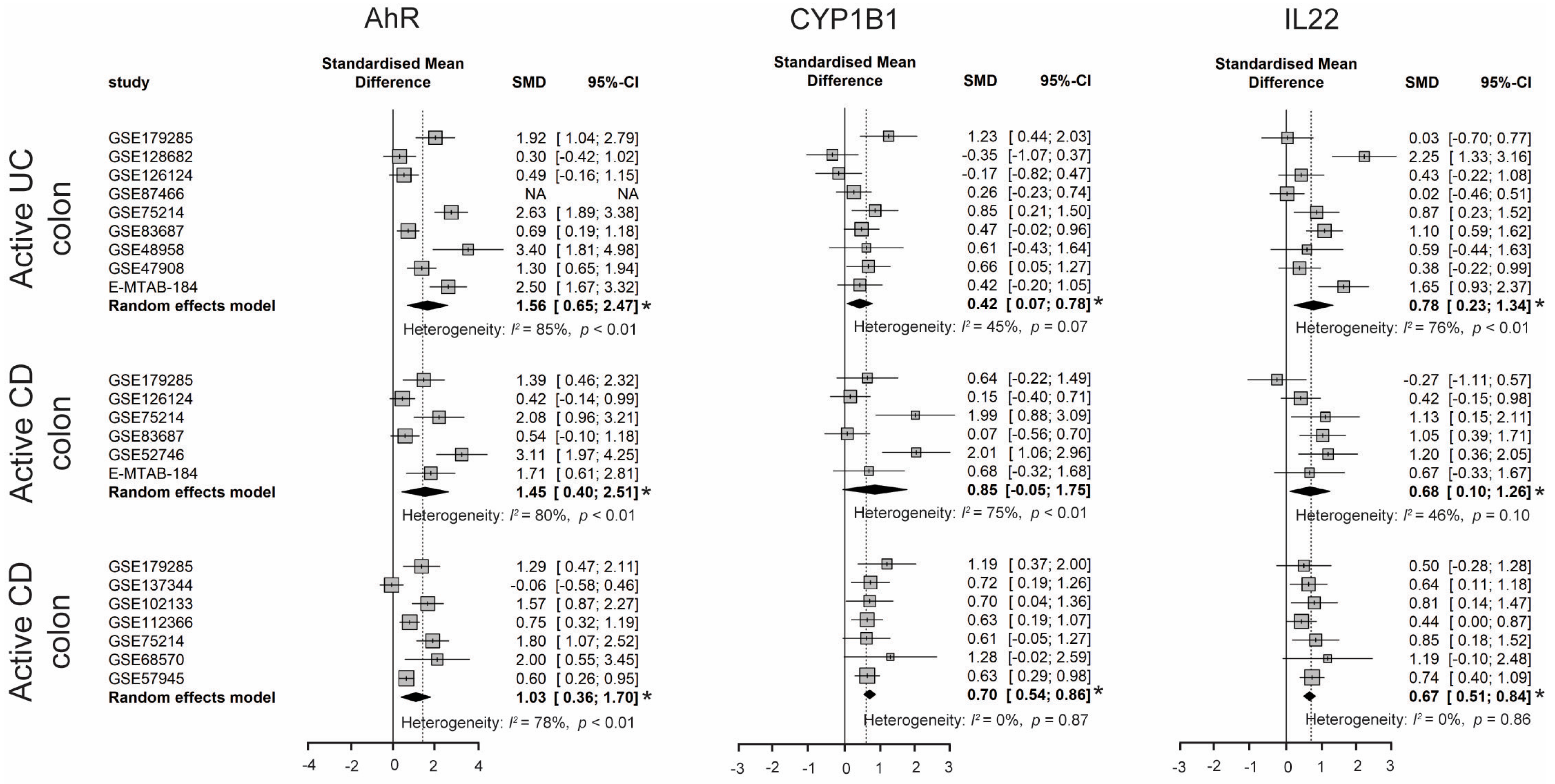
3.2.5. Activated AhR Signaling in IBD Patients
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef]
- Law, C.C.Y.; Sasidharan, S.; Rodrigues, R.; Nguyen, D.D.; Sauk, J.; Garber, J.; Giallourakis, C.; Xavier, R.; Khalili, H.; Yajnik, V.; et al. Impact of Specialized Inpatient IBD Care on Outcomes of IBD Hospitalizations: A Cohort Study. Inflamm. Bowel Dis. 2016, 22, 2149–2157. [Google Scholar] [CrossRef]
- Kato, J.; Yoshida, T.; Hiraoka, S. Prediction of treatment outcome and relapse in inflammatory bowel disease. Expert Rev. Clin. Immunol. 2019, 15, 667–677. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef]
- Kaluzna-Czaplinska, J.; Gatarek, P.; Chirumbolo, S.; Chartrand, M.S.; Bjorklund, G. How important is tryptophan in human health? Crit. Rev. Food Sci. 2019, 59, 72–88. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Keszthelyi, D.; Troost, F.J.; Masclee, A.A. Understanding the role of tryptophan and serotonin metabolism in gastrointestinal function. Neurogastroenterol. Motil. 2009, 21, 1239–1249. [Google Scholar] [CrossRef]
- Sun, M.; Ma, N.; He, T.; Johnston, L.J.; Ma, X. Tryptophan (Trp) modulates gut homeostasis via aryl hydrocarbon receptor (AhR). Crit. Rev. Food Sci. Nutr. 2020, 60, 1760–1768. [Google Scholar] [CrossRef]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Islam, J.; Sato, S.; Watanabe, K.; Watanabe, T.; Ardiansyah; Hirahara, K.; Aoyama, Y.; Tomita, S.; Aso, H.; Komai, M.; et al. Dietary tryptophan alleviates dextran sodium sulfate-induced colitis through aryl hydrocarbon receptor in mice. J. Nutr. Biochem. 2017, 42, 43–50. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D'Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan Catabolites from Microbiota Engage Aryl Hydrocarbon Receptor and Balance Mucosal Reactivity via Interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Wlodarska, M.; Luo, C.W.; Kolde, R.; d’Hennezel, E.; Annand, J.W.; Heim, C.E.; Krastel, P.; Schmitt, E.K.; Omar, A.S.; Creasey, E.A.; et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe 2017, 22, 25–37. [Google Scholar] [CrossRef]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Hasler, R.; et al. Increased Tryptophan Metabolism Is Associated with Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516. [Google Scholar] [CrossRef]
- Sofia, M.A.; Ciorba, M.A.; Meckel, K.; Lim, C.K.; Guillemin, G.J.; Weber, C.R.; Bissonnette, M.; Pekow, J.R. Tryptophan Metabolism through the Kynurenine Pathway is Associated with Endoscopic Inflammation in Ulcerative Colitis. Inflamm. Bowel Dis. 2018, 24, 1471–1480. [Google Scholar] [CrossRef]
- Gupta, N.K.; Thaker, A.I.; Kanuri, N.; Riehl, T.E.; Rowley, C.W.; Stenson, W.F.; Ciorba, M.A. Serum analysis of tryptophan catabolism pathway: Correlation with Crohn’s disease activity. Inflamm. Bowel Dis. 2012, 18, 1214–1220. [Google Scholar] [CrossRef]
- Ghia, J.E.; Li, N.; Wang, H.Q.; Collins, M.; Deng, Y.K.; El-Sharkawy, R.T.; Cote, F.; Mallet, J.; Khan, W.I. Serotonin Has a Key Role in Pathogenesis of Experimental Colitis. Gastroenterology 2009, 137, 1649–1660. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. PLoS Med. 2021, 18, e1003583. [Google Scholar] [CrossRef]
- Harrer, M.; Cuijpers, P.; Furukawa, T.A.; Ebert, D.D. Doing Meta-Analysis with R: A Hands-On Guide; Chapman & Hall/CRC Press (Taylor & Francis): New York, NY, USA, 2021. [Google Scholar]
- Schwarzer, G.; Carpenter, J.R.; Rücker, G. Meta-Analysis with R; Springer: Heidelberg, Germany, 2015. [Google Scholar]
- Di’Narzo, A.F.; Houten, S.M.; Kosoy, R.; Huang, R.Q.; Vaz, F.M.; Hou, R.X.; Wei, G.; Wang, W.H.; Comella, P.H.; Dodatko, T.; et al. Integrative Analysis of the Inflammatory Bowel Disease Serum Metabolome Improves Our Understanding of Genetic Etiology and Points to Novel Putative Therapeutic Targets. Gastroenterology 2022, 162, 828–843. [Google Scholar] [CrossRef]
- Gu, Z.; Pei, W.; Shen, Y.; Wang, L.; Zhu, J.; Zhang, Y.; Fan, S.; Wu, Q.; Li, L.; Zhang, Z. Akkermansia muciniphila and its outer protein Amuc_1100 regulates tryptophan metabolism in colitis. Food Funct. 2021, 12, 10184–10195. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, X.; Zhang, X.; Xiao, F.; Hu, H.; Li, X.; Dong, F.; Sun, M.; Xiao, Y.; Ge, T.; et al. Microbial and metabolic features associated with outcome of infliximab therapy in pediatric Crohn’s disease. Gut Microbes 2021, 13, 1865708. [Google Scholar] [CrossRef]
- Notararigo, S.; Martin-Pastor, M.; Vinuela-Roldan, J.E.; Quiroga, A.; Dominguez-Munoz, J.E.; Barreiro-de Acosta, M. Targeted 1H NMR metabolomics and immunological phenotyping of human fresh blood and serum samples discriminate between healthy individuals and inflammatory bowel disease patients treated with anti-TNF. J. Mol. Med. 2021, 99, 1251–1264. [Google Scholar] [CrossRef]
- Huhn, M.; Juan, M.H.S.; Melcher, B.; Dreis, C.; Schmidt, K.G.; Schwiebs, A.; Collins, J.; Pfeilschifter, J.M.; Vieth, M.; Stein, J.; et al. Inflammation-Induced Mucosal KYNU Expression Identifies Human Ileal Crohn’s Disease. J. Clin. Med. 2020, 9, 1360. [Google Scholar] [CrossRef]
- Manzella, C.R.; Jayawardena, D.; Pagani, W.; Li, Y.; Alrefai, W.A.; Bauer, J.; Jung, B.; Weber, C.R.; Gill, R.K. Serum Serotonin Differentiates Between Disease Activity States in Crohn’s Patients. Inflamm. Bowel Dis. 2020, 26, 1607–1618. [Google Scholar] [CrossRef]
- Diab, J.; Hansen, T.; Goll, R.; Stenlund, H.; Jensen, E.; Moritz, T.; Florholmen, J.; Forsdahl, G. Mucosal Metabolomic Profiling and Pathway Analysis Reveal the Metabolic Signature of Ulcerative Colitis. Metabolites 2019, 9, 291. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Lai, Y.; Xue, J.; Liu, C.W.; Gao, B.; Chi, L.; Tu, P.; Lu, K.; Ru, H. Serum Metabolomics Identifies Altered Bioenergetics, Signaling Cascades in Parallel with Exposome Markers in Crohn’s Disease. Molecules 2019, 24, 449. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Whiley, L.; Nye, L.C.; Grant, I.; Andreas, N.; Chappell, K.E.; Sarafian, M.H.; Misra, R.; Plumb, R.S.; Lewis, M.R.; Nicholson, J.K.; et al. Ultrahigh-Performance Liquid Chromatography Tandem Mass Spectrometry with Electrospray Ionization Quantification of Tryptophan Metabolites and Markers of Gut Health in Serum and Plasma-Application to Clinical and Epidemiology Cohorts. Anal. Chem. 2019, 91, 5207–5216. [Google Scholar] [CrossRef]
- Shajib, M.S.; Chauhan, U.; Adeeb, S.; Chetty, Y.; Armstrong, D.; Halder, S.L.S.; Marshall, J.K.; Khan, W.I. Characterization of Serotonin Signaling Components in Patients with Inflammatory Bowel Disease. J. Can. Assoc. Gastroenterol. 2019, 2, 132–140. [Google Scholar] [CrossRef]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef]
- Bosch, S.; Struys, E.A.; van Gaal, N.; Bakkali, A.; Jansen, E.W.; Diederen, K.; Benninga, M.A.; Mulder, C.J.; de Boer, N.K.H.; de Meij, T.G.J. Fecal Amino Acid Analysis Can Discriminate De Novo Treatment-Naive Pediatric Inflammatory Bowel Disease from Controls. J. Pediatr. Gastr. Nutr. 2018, 66, 773–778. [Google Scholar] [CrossRef]
- Scoville, E.A.; Allaman, M.M.; Brown, C.T.; Motley, A.K.; Horst, S.N.; Williams, C.S.; Koyama, T.; Zhao, Z.; Adams, D.W.; Beaulieu, D.B.; et al. Alterations in Lipid, Amino Acid, and Energy Metabolism Distinguish Crohn’s Disease from Ulcerative Colitis and Control Subjects by Serum Metabolomic Profiling. Metabolomics 2018, 14, 17. [Google Scholar] [CrossRef]
- Abautret-Daly, Á.; Dempsey, E.; Riestra, S.; de Francisco-García, R.; Parra-Blanco, A.; Rodrigo, L.; Medina, C.; Connor, T.J.; Harkin, A. Association between psychological measures with inflammatory anddisease-related markers of inflammatory bowel disease. Int. J. Psychiatry Clin. Pract. 2017, 21, 221–230. [Google Scholar] [CrossRef]
- Kolho, K.L.; Pessia, A.; Jaakkola, T.; de Vos, W.M.; Velagapudi, V. Faecal and Serum Metabolomics in Paediatric Inflammatory Bowel Disease. J. Crohn’s Colitis 2017, 11, 321–334. [Google Scholar] [CrossRef]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.Y.; Huang, S.G.; Zhang, H.Y.; Ye, H.; Chi, H.G.; Zou, Y.; Lv, R.X.; Zheng, X.B. Comparison of 5-hydroxytryptophan signaling pathway characteristics in diarrhea-predominant irritable bowel syndrome and ulcerative colitis. World J. Gastroenterol. 2016, 22, 3451–3459. [Google Scholar] [CrossRef]
- De Preter, V.; Machiels, K.; Joossens, M.; Arijs, I.; Matthys, C.; Vermeire, S.; Rutgeerts, P.; Verbeke, K. Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut 2015, 64, 447–458. [Google Scholar] [CrossRef]
- Kohashi, M.; Nishiumi, S.; Ooi, M.; Yoshie, T.; Matsubara, A.; Suzuki, M.; Hoshi, N.; Kamikozuru, K.; Yokoyama, Y.; Fukunaga, K.; et al. A novel gas chromatography mass spectrometry-based serum diagnostic and assessment approach to ulcerative colitis. J. Crohn’s Colitis 2014, 8, 1010–1021. [Google Scholar] [CrossRef]
- Yau, Y.Y.; Leong, R.W.; Shin, S.; Bustamante, S.; Pickford, R.; Hejazi, L.; Campbell, B.; Wasinger, V.C. Bimodal plasma metabolomics strategy identifies novel inflammatory metabolites in inflammatory bowel diseases. Discov. Med. 2014, 18, 113–124. [Google Scholar]
- Walton, C.; Fowler, D.P.; Turner, C.; Jia, W.; Whitehead, R.N.; Griffiths, L.; Dawson, C.; Waring, R.H.; Ramsden, D.B.; Cole, J.A.; et al. Analysis of volatile organic compounds of bacterial origin in chronic gastrointestinal diseases. Inflamm. Bowel Dis. 2013, 19, 2069–2078. [Google Scholar] [CrossRef]
- Hisamatsu, T.; Okamoto, S.; Hashimoto, M.; Muramatsu, T.; Andou, A.; Uo, M.; Kitazume, M.T.; Matsuoka, K.; Yajima, T.; Inoue, N.; et al. Novel, objective, multivariate biomarkers composed of plasma amino acid profiles for the diagnosis and assessment of inflammatory bowel disease. PLoS ONE 2012, 7, e31131. [Google Scholar] [CrossRef] [PubMed]
- Ooi, M.; Nishiumi, S.; Yoshie, T.; Shiomi, Y.; Kohashi, M.; Fukunaga, K.; Nakamura, S.; Matsumoto, T.; Hatano, N.; Shinohara, M.; et al. GC/MS-based profiling of amino acids and TCA cycle-related molecules in ulcerative colitis. Inflamm. Res. 2011, 60, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef]
- Rossi, F.; Miggiano, R.; Ferraris, D.M.; Rizzi, M. The Synthesis of Kynurenic Acid in Mammals: An Updated Kynurenine Aminotransferase Structural KATalogue. Front. Mol. Biosci. 2019, 6, 7. [Google Scholar] [CrossRef]
- Badawy, A.A. Hypothesis kynurenic and quinolinic acids: The main players of the kynurenine pathway and opponents in inflammatory disease. Med. Hypotheses 2018, 118, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Namboodiri, M.A. Tryptophan and the immune response. Immunol. Cell Biol. 2003, 81, 247–265. [Google Scholar] [CrossRef]
- Peters, J.C. Tryptophan Nutrition and Metabolism—An Overview. Adv. Exp. Med. Biol. 1991, 294, 345–358. [Google Scholar]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf9794. [Google Scholar] [CrossRef]
- Mandi, Y.; Vecsei, L. The kynurenine system and immunoregulation. J. Neural Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef]
- Ostapiuk, A.; Urbanska, E.M. Kynurenic acid in neurodegenerative disorders-unique neuroprotection or double-edged sword? Cns. Neurosci. Ther. 2022, 28, 19–35. [Google Scholar] [CrossRef]
- Kaszaki, J.; Erces, D.; Varga, G.; Szabo, A.; Vecsei, L.; Boros, M. Kynurenines and intestinal neurotransmission: The role of N-methyl-d-aspartate receptors. J. Neural Transm. 2012, 119, 211–223. [Google Scholar] [CrossRef]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.L.; Bohorquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361, eaat5236. [Google Scholar] [CrossRef]
- Zheng, H.; Teague, T.K.; Yeh, F.C.; Burrows, K.; Figueroa-Hall, L.K.; Aupperle, R.L.; Khalsa, S.S.; Paulus, M.P.; Savitz, J. C-Reactive protein and the kynurenic acid to quinolinic acid ratio are independently associated with white matter integrity in major depressive disorder. Brain Behav. Immun. 2022, 105, 180–189. [Google Scholar] [CrossRef]
- Savitz, J.; Drevets, W.C.; Wurfel, B.E.; Ford, B.N.; Bellgowan, P.S.F.; Victor, T.A.; Bodurka, J.; Teague, T.K.; Dantzer, R. Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain Behav. Immun. 2015, 46, 55–59. [Google Scholar] [CrossRef]
- Tao, E.; Zhu, Z.; Hu, C.; Long, G.; Chen, B.; Guo, R.; Fang, M.; Jiang, M. Potential Roles of Enterochromaffin Cells in Early Life Stress-Induced Irritable Bowel Syndrome. Front. Cell. Neurosci. 2022, 16, 837166. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Grondin, J.A.; Khan, W.I. Tryptophan-derived serotonin-kynurenine balance in immune activation and intestinal inflammation. FASEB J. 2021, 35, e21888. [Google Scholar] [CrossRef]
- Banskota, S.; Ghia, J.E.; Khan, W.I. Serotonin in the gut: Blessing or a curse. Biochimie 2019, 161, 56–64. [Google Scholar] [CrossRef]
- Liu, N.; Sun, S.; Wang, P.; Sun, Y.; Hu, Q.; Wang, X. The Mechanism of Secretion and Metabolism of Gut-Derived 5-Hydroxytryptamine. Int. J. Mol. Sci. 2021, 22, 7931. [Google Scholar] [CrossRef]
- Mawe, G.M.; Hoffman, J.M. Serotonin signalling in the gut-functions, dysfunctions and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 473–486. [Google Scholar] [CrossRef]
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int. J. Mol. Sci. 2021, 22, 2973. [Google Scholar] [CrossRef]
- Kim, J.J.; Wang, H.Q.; Terc, J.D.; Zambrowicz, B.; Yang, Q.M.; Khan, W.I. Blocking peripheral serotonin synthesis by telotristat etiprate (LX1032/LX1606) reduces severity of both chemical- and infection-induced intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G455–G465. [Google Scholar] [CrossRef]
- Spohn, S.N.; Bianco, F.; Scott, R.B.; Keenan, C.M.; Linton, A.A.; O’Neill, C.H.; Bonora, E.; Dicay, M.; Lavoie, B.; Wilcox, R.L.; et al. Protective Actions of Epithelial 5-Hydroxytryptamine 4 Receptors in Normal and Inflamed Colon. Gastroenterology 2016, 151, 933–944.e3. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wichems, C.; Heils, A.; Lesch, K.P.; Murphy, D.L. Reduction in the density and expression, but not G-protein coupling, of serotonin receptors (5-HT1A) in 5-HT transporter knock-out mice: Gender and brain region differences. J. Neurosci. 2000, 20, 7888–7895. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef]
- Su, X.; Gao, Y.; Yang, R. Gut Microbiota-Derived Tryptophan Metabolites Maintain Gut and Systemic Homeostasis. Cells 2022, 11, 2296. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 2010, 34, 426–444. [Google Scholar] [CrossRef]
- Chimerel, C.; Emery, E.; Summers, D.K.; Keyser, U.; Gribble, F.M.; Reimann, F. Bacterial metabolite indole modulates incretin secretion from intestinal enteroendocrine L cells. Cell Rep. 2014, 9, 1202–1208. [Google Scholar] [CrossRef]
- Williams, B.B.; Van Benschoten, A.H.; Cimermancic, P.; Donia, M.S.; Zimmermann, M.; Taketani, M.; Ishihara, A.; Kashyap, P.C.; Fraser, J.S.; Fischbach, M.A. Discovery and Characterization of Gut Microbiota Decarboxylases that Can Produce the Neurotransmitter Tryptamine. Cell Host Microbe 2014, 16, 495–503. [Google Scholar] [CrossRef]
- Bhattarai, Y.; Jie, S.; Linden, D.R.; Ghatak, S.; Mars, R.A.T.; Williams, B.B.; Pu, M.; Sonnenburg, J.L.; Fischbach, M.A.; Farrugia, G.; et al. Bacterially Derived Tryptamine Increases Mucus Release by Activating a Host Receptor in a Mouse Model of Inflammatory Bowel Disease. iScience 2020, 23, 101798. [Google Scholar] [CrossRef]
- Bhattarai, Y.; Williams, B.B.; Battaglioli, E.J.; Whitaker, W.R.; Till, L.; Grover, M.; Linden, D.R.; Akiba, Y.; Kandimalla, K.K.; Zachos, N.C.; et al. Gut Microbiota-Produced Tryptamine Activates an Epithelial G-Protein-Coupled Receptor to Increase Colonic Secretion. Cell Host Microbe 2018, 23, 775–785.e5. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef]
- Vyhlidalova, B.; Krasulova, K.; Pecinkova, P.; Marcalikova, A.; Vrzal, R.; Zemankova, L.; Vanco, J.; Travnicek, Z.; Vondracek, J.; Karasova, M.; et al. Gut Microbial Catabolites of Tryptophan Are Ligands and Agonists of the Aryl Hydrocarbon Receptor: A Detailed Characterization. Int. J. Mol. Sci. 2020, 21, 2614. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug. Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef]
- Manzella, C.; Singhal, M.; Alrefai, W.A.; Saksena, S.; Dudeja, P.K.; Gill, R.K. Serotonin is an endogenous regulator of intestinal CYP1A1 via AhR. Sci. Rep. 2018, 8, 6103. [Google Scholar] [CrossRef]
- Wyatt, M.; Greathouse, K.L. Targeting Dietary and Microbial Tryptophan-Indole Metabolism as Therapeutic Approaches to Colon Cancer. Nutrients 2021, 13, 1189. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, K.; Han, B.; Sheng, B.; Yin, J.; Pu, A.; Li, L.; Sun, L.; Yu, M.; Qiu, Y.; et al. Aryl hydrocarbon receptor inhibits inflammation in DSS-induced colitis via the MK2/p-MK2/TTP pathway. Int. J. Mol. Med. 2018, 41, 868–876. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Lamas, B.; Natividad, J.M.; Sokol, H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol. 2018, 11, 1024–1038. [Google Scholar] [CrossRef]
- Vondracek, J.; Umannova, L.; Machala, M. Interactions of the Aryl Hydrocarbon Receptor with Inflammatory Mediators: Beyond CYP1A Regulation. Curr. Drug Metab. 2011, 12, 89–103. [Google Scholar] [CrossRef]
- Arsenescu, R.; Arsenescu, V.; Zhong, J.A.; Nasser, M.; Melinte, R.; Dingle, R.W.C.; Swanson, H.; de Villiers, W.J. Role of the Xenobiotic Receptor in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2011, 17, 1149–1162. [Google Scholar] [CrossRef]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl Hydrocarbon Receptor-Induced Signals Up-regulate IL-22 Production and Inhibit Inflammation in the Gastrointestinal Tract. Gastroenterology 2011, 141, 237–248.e1. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Function | Gene Symbols |
|---|---|
| Tryptophan absorption | SLC6A19, SLC16A10 |
| Kynurenine pathway | IDO1, AFMID, KYAT1, KYAT2, KMO, KYNU, HAAO, ACMSD |
| Melatonin biosynthesis | TPH1, AADC, AANAT, ASMT * |
| Serotonin transporter | SLC6A4 |
| Serotonin metabolism enzyme | MAOA, MAOB |
| Serotonin (5-HT) receptors | 5-HTR1A *, 5-HTR1B, 5-HTR1C *, 5-HTR1D, 5-HTR1E, 5-HTR1F, 5-HTR2A, 5-HTR2B, 5-HTR2C *, 5-HTR3A, 5-HTR3B, 5-HTR3C, 5-HTR3D *, 5-HTR3E, 5-HTR4, 5-HTR5A *, 5-HTR5B *, 5-HTR6, 5-HTR7 |
| AhR signaling pathway | AhR, ARNT, AHRR *, CYP1A1, CYP1B1, IL22 |
| GEO ID/ ArrayExpress ID (Publication Year) | Sample Size after Removing Outliers | Inflammation Status of IBD Patients | Platform | Adult/ Pediatric Cohort | ||
|---|---|---|---|---|---|---|
| Disease | Control | Active | Inactive | |||
| CD vs. Control (ileum) | ||||||
| GSE179285 (2021) | 62 | 8 | 33 | 29 | Agilent-014850 Whole Human Genome Microarray 4 × 44k G4112F (GPL6480) | Adult |
| GSE137344 (2020) | 99 | 29 | 28 | 71 | NextSeq 550 (GPL21697) | Pediatric |
| GSE102133 (2019) | 54 | 11 | 54 | 0 | Affymetrix human gene 1.0 ST Array (GPL6244) | Adult |
| GSE112366 (2019) | 105 | 26 | 105 | 0 | Affymetrix HT HG-U133 + PM Array Plate (GPL13158) | Adult |
| E-MTAB-5790 (2018) | 36 | 32 | 0 | 36 | Agilent Whole Human Genome 4 × 44k Microarray | Pediatric |
| GSE75214 (2017) | 67 | 11 | 51 | 16 | Affymetrix Human Gene 1.0 ST Array (GPL6244) | Adult |
| GSE68570 (2016) | 6 | 5 | 6 | 0 | Illumina HumanHT-12 V4.0 expression BeadChip (GPL10558) | Adult |
| GSE57945 (2014) | 163 | 42 | 163 # | 0 | Illumina HiSeq 2000 (GPL11154) | Pediatric |
| CD vs. Control (colon) | ||||||
| GSE179285 (2021) | 71 | 11 | 11 | 60 | Agilent-014850 Whole Human Genome Microarray 4 × 44k G4112F (GPL6480) | Adult |
| GSE126124 (2019) | 38 | 19 | 38 # | 0 | Affymetrix human gene 1.0 ST Array (GPL6244) | Pediatric |
| GSE75214 (2017) | 8 | 11 | 8 | 0 | Affymetrix Human Gene 1.0 ST Array (GPL6244) | Adult |
| GSE83687 (2017) | 12 | 48 | 12 | 0 | Illumina HiSeq 2500 (GPL16791) | Pediatric and adult |
| GSE52746 (2014) | 10 | 17 | 10 | 0 | Affymetrix Human Genome U133 Plus 2.0 Array (GPL570) | Adult |
| E-MTAB-184 (2012) | 24 | 19 | 5 | 19 | Illumina HumanHT-12 v3.0 Expression BeadChip | Adult |
| UC vs. Control (colon) | ||||||
| GSE179285 (2021) | 40 | 11 | 20 | 20 | Agilent-014850 Whole Human Genome Microarray 4 × 44k G4112F (GPL6480) | Adult |
| GSE128682 (2020) | 28 | 16 | 14 | 14 | NextSeq 550 (GPL21697) | Adult |
| GSE126124 (2019) | 18 | 19 | 18 # | 0 | Affymetrix human gene 1.0 ST Array (GPL6244) | Pediatric |
| GSE87466 (2018) | 86 | 20 | 86 # | 0 | Affymetrix HT HG-U133 + PM Array Plate (GPL13158) | Adult |
| GSE75214 (2017) | 97 | 11 | 74 | 23 | Affymetrix Human Gene 1.0 ST Array (GPL6244) | Adult |
| GSE83687 (2017) | 25 | 48 | 25 | 0 | Illumina HiSeq 2500 (GPL16791) | Pediatric and adult |
| GSE48958 (2015) | 13 | 8 | 7 | 6 | Affymetrix human gene 1.0 ST Array (GPL6244) | Adult |
| GSE47908 (2014) | 38 | 15 | 38 | 0 | Affymetrix Human Genome U133 Plus 2.0 Array (GPL570) | Adult |
| E-MTAB-184 (2012) | 61 | 19 | 21 | 40 | Illumina HumanHT-12 v3.0 Expression BeadChip | Adult |
| Study (Publication Year) | Patients with CD | Patients with UC | Control | Biosample | Analytical Technique | ||
|---|---|---|---|---|---|---|---|
| Active | Inactive | Active | Inactive | ||||
| Di’Narzo et al. (2022) [20] | 284 * | 360 | 329 | Serum | LC-MS | ||
| 88 | 101 | 72 | Stool | ||||
| Gu et al. (2021) [21] | / | / | 93 | / | 102 | Serum | LC-MS |
| Wang et al. (2021) [22] | 29 | / | / | / | 20 | Stool | LC-MS |
| Notararigo et al. (2021) [23] | / | 18 | / | 9 | 10 | Serum | 1H-NMR |
| Huhn et al. (2020) [24] | 12 | / | 11 | / | 12 | Intestinal biopsy | Immunohistochemistry |
| Manzella et al. (2020) [25] | 15 | 15 | 15 | 15 | / | Serum | LC-MS |
| Diab et al. (2019) [26] | / | / | 18 | 10 | 14 | Intestinal biopsy | GC-MS LC-MS |
| Franzosa et al. (2019) [27] | 68 | 53 | 34 | Stool | LC-MS | ||
| Lai et al. (2019) [28] | 10 | 10 | / | / | 10 | Serum | LC-MS |
| Lloyd-Price et al. (2019) [29] | 67 | 38 | 27 | Stool | LC-MS | ||
| Whiley et al. (2019) [30] | / | / | 19 | 10 | Plasma | LC-MS | |
| Shajib et al. (2019) [31] | 21 | 19 | / | / | 40 | Plasma | ELISA |
| Alexeev et al. (2018) [32] | / | / | 15 | 20 | 20 | Serum | HPLC-electrochemical coulometric array |
| Bosch et al. (2018) [33] | 15 | / | 15 | / | 15 | Stool | HPLC-UV |
| Scoville et al. (2018) [34] | 8 | 12 | 19 | 1 | 20 | Serum | LC-MS |
| Abautret-Daly et al. (2017) [35] | 8 | 10 | 19 | Plasma | HPLC coupled PDA-UV and fluorescence detectors | ||
| Kolho et al. (2017) [36] | 36 | / | 20 | / | 29 | Serum and stool | LC-MS |
| Nikolaus et al. (2017) [13] | 81 | 67 | 100 | Serum | LC-MS | ||
| Lamas et al. (2016) [37] | 54 # | 32 | Stool | HPLC-coulometric electrode assay LC-MS | |||
| Yu et al. (2016) [38] | / | / | / | 33 | 30 | Plasma | HPLC-electrochemical detector |
| De Preter et al. (2015) [39] | 29 | 54 | 28 | 40 | 40 | Stool | GC-MS |
| Kohashi et al. (2014) [40] | / | / | 52 | 68 | 120 | Serum | GC-MS |
| Yau et al. (2014) [41] | 15 | 10 | 14 | 5 | 9 | Plasma | GC-MS |
| Walton et al. (2013) [42] | 22 | / | 20 | / | 19 | Stool | GC-MS |
| Hisamatsu et al. (2012) [43] | 29 | 73 | 38 | 64 | 102 | Plasma | HPLC-spectrophotometry |
| Gupta et al. (2012) [15] | 20 | 5 | / | / | 11 | Serum | HPLC coupled UV-V detector |
| Ooi et al. (2011) [44] | 16 | 5 | 1 | 12 | 17 | Serum | GC-MS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; van Schooten, F.-J.; Jin, H.; Jonkers, D.; Godschalk, R. The Involvement of Intestinal Tryptophan Metabolism in Inflammatory Bowel Disease Identified by a Meta-Analysis of the Transcriptome and a Systematic Review of the Metabolome. Nutrients 2023, 15, 2886. https://doi.org/10.3390/nu15132886
Wang S, van Schooten F-J, Jin H, Jonkers D, Godschalk R. The Involvement of Intestinal Tryptophan Metabolism in Inflammatory Bowel Disease Identified by a Meta-Analysis of the Transcriptome and a Systematic Review of the Metabolome. Nutrients. 2023; 15(13):2886. https://doi.org/10.3390/nu15132886
Chicago/Turabian StyleWang, Shan, Frederik-Jan van Schooten, Han Jin, Daisy Jonkers, and Roger Godschalk. 2023. "The Involvement of Intestinal Tryptophan Metabolism in Inflammatory Bowel Disease Identified by a Meta-Analysis of the Transcriptome and a Systematic Review of the Metabolome" Nutrients 15, no. 13: 2886. https://doi.org/10.3390/nu15132886
APA StyleWang, S., van Schooten, F. -J., Jin, H., Jonkers, D., & Godschalk, R. (2023). The Involvement of Intestinal Tryptophan Metabolism in Inflammatory Bowel Disease Identified by a Meta-Analysis of the Transcriptome and a Systematic Review of the Metabolome. Nutrients, 15(13), 2886. https://doi.org/10.3390/nu15132886