Microbial Diversity and Toxin Risk in Tropical Freshwater Reservoirs of Cape Verde


Abstract
:1. Introduction
2. Results
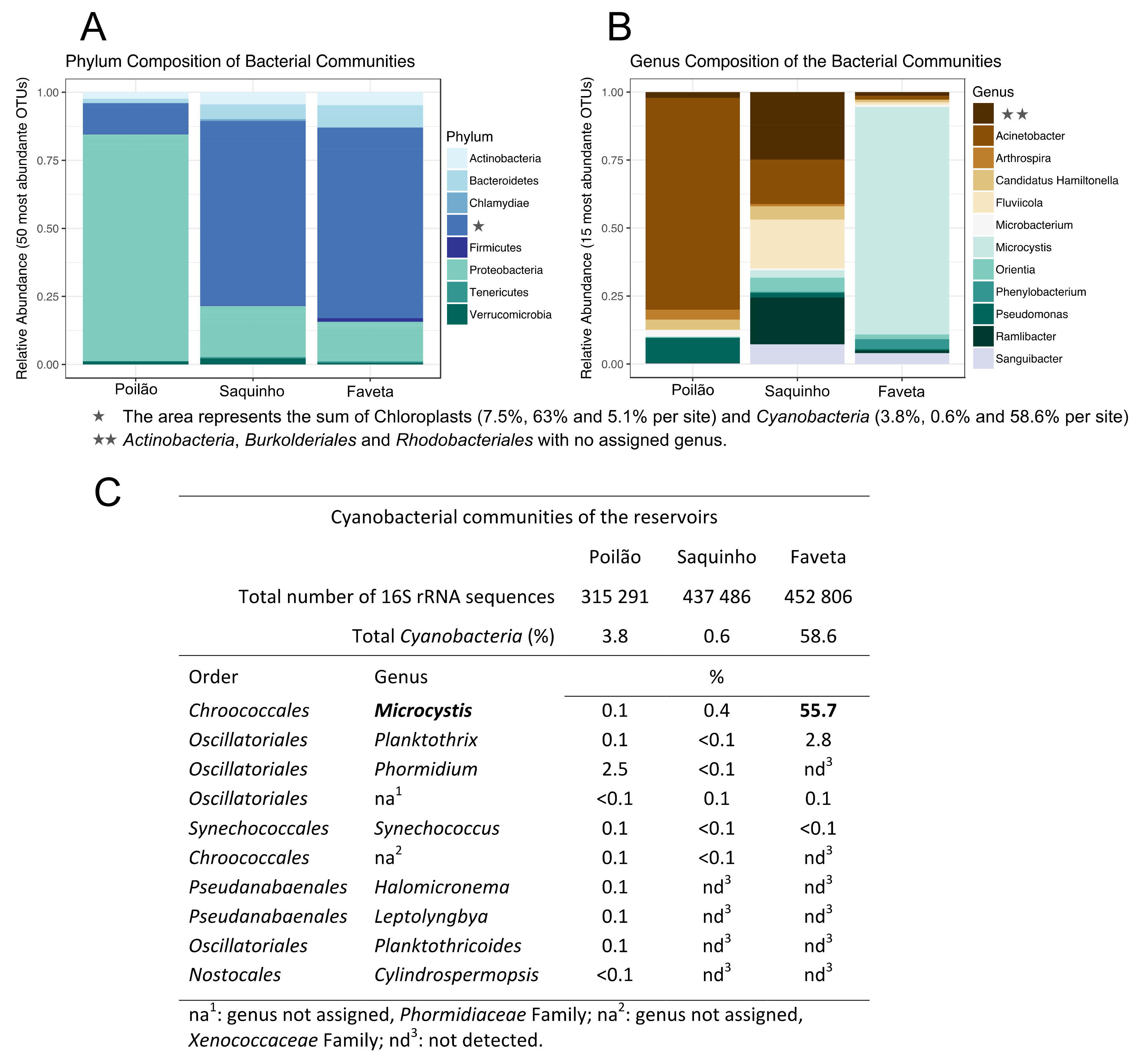
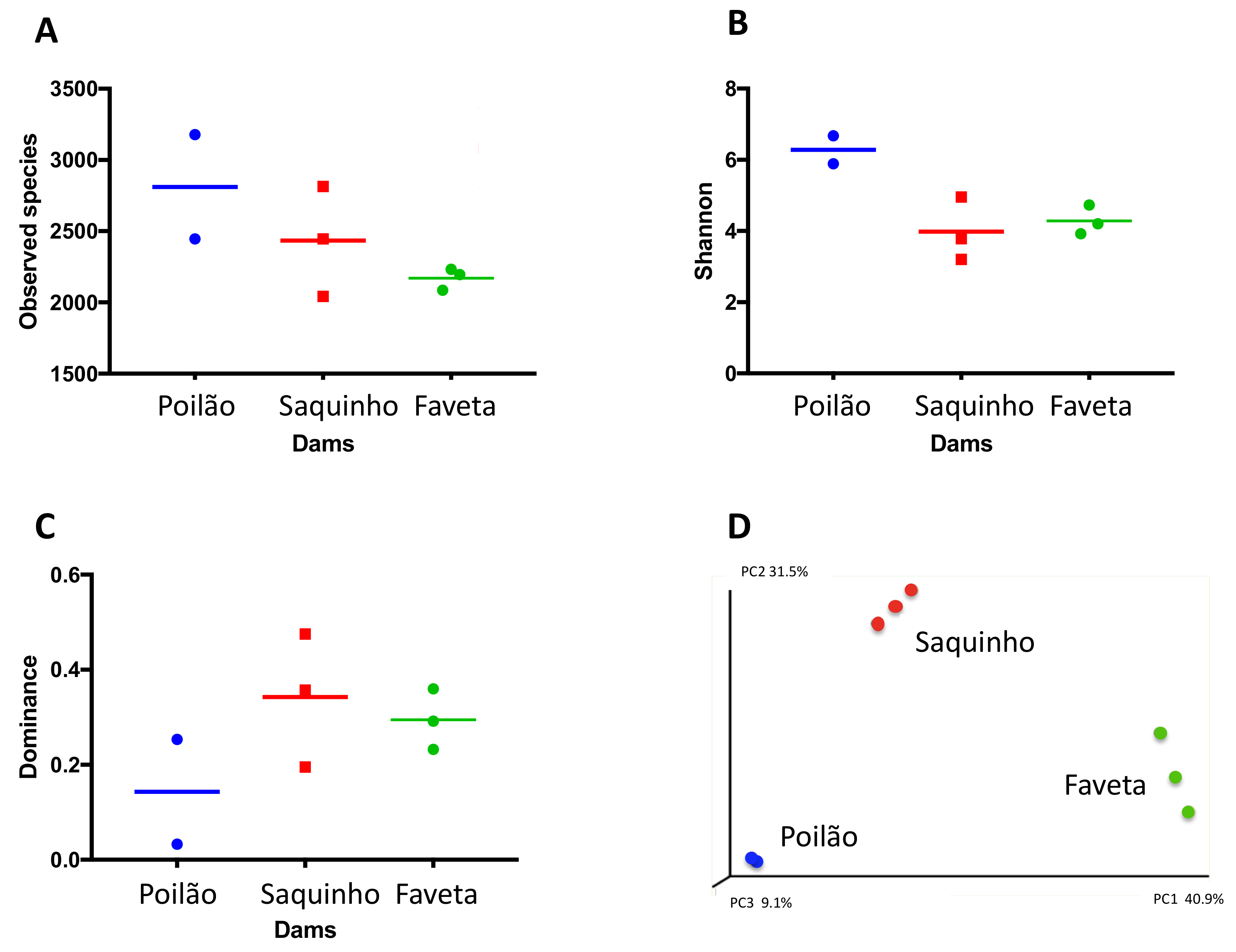
2.1. Diversity of the Microbial Communities in the Reservoirs
2.2. Toxin Producing Genera/Species
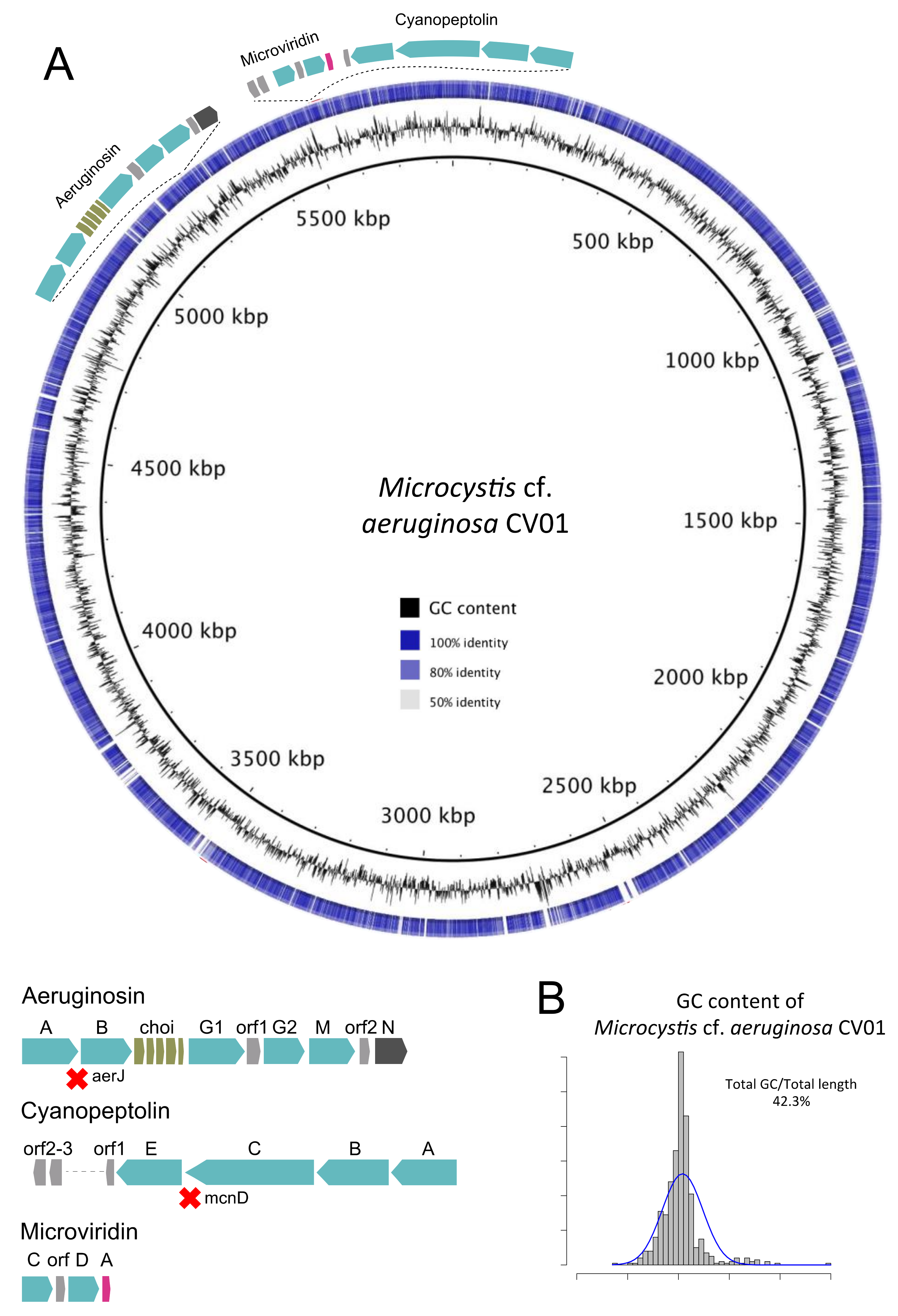
2.3. The Presence of a Dominant Mycrocystis Species in Faveta: Reconstruction of M. cf. aeruginosa CV01 Genome
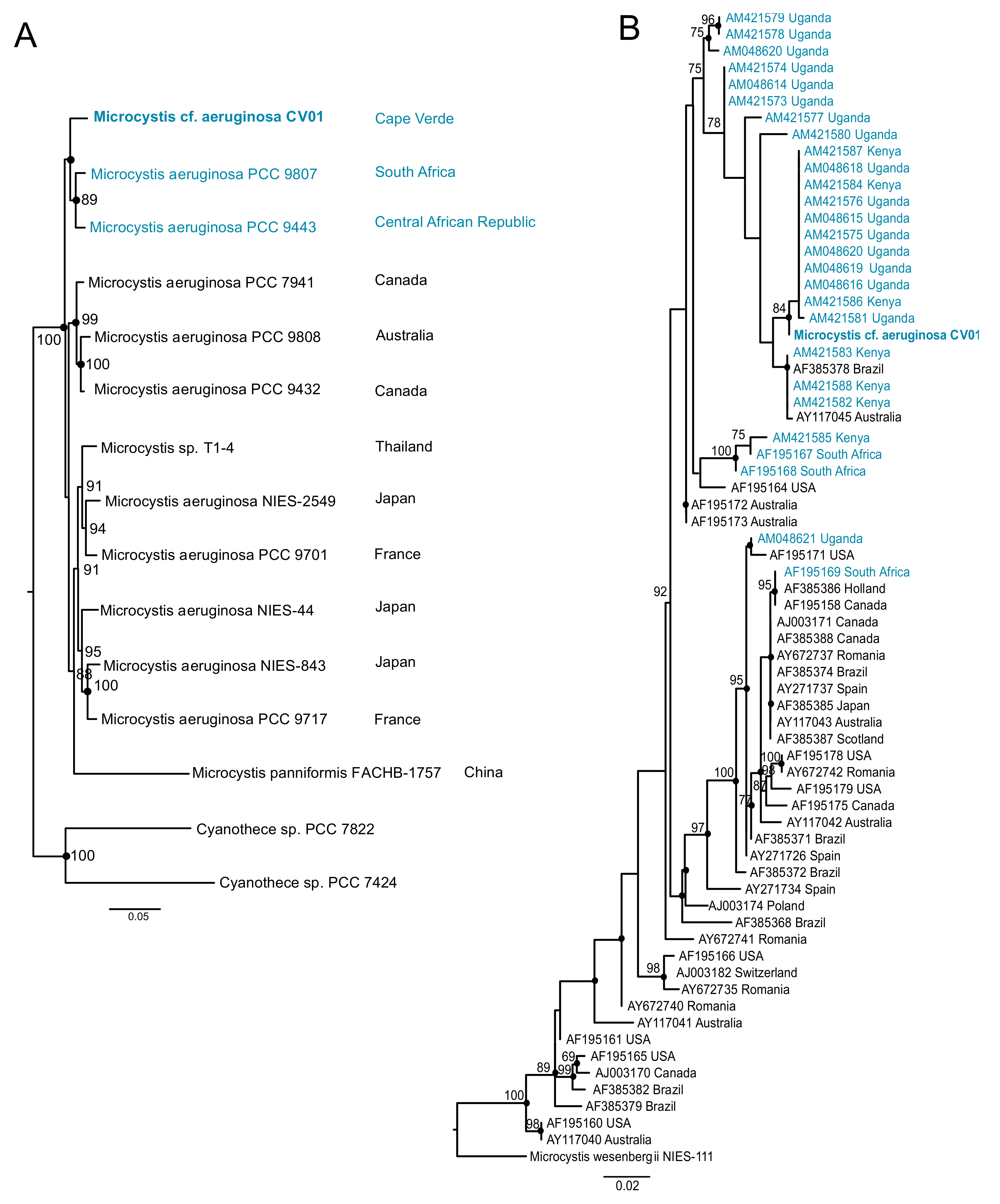
2.4. Phylogenetic Analysis
2.5. Toxin Genes and Toxic Species
3. Discussion
4. Materials and Methods
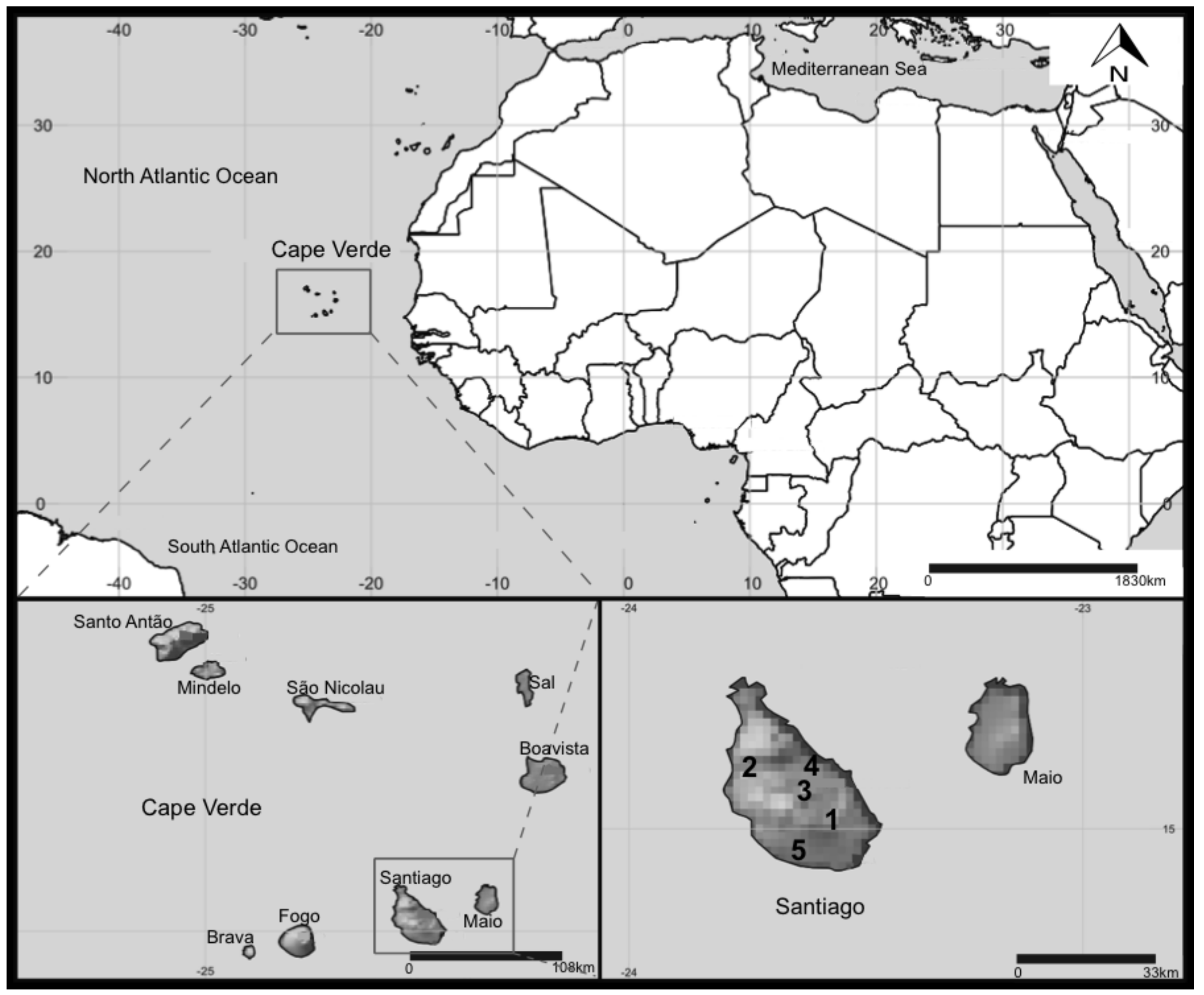
4.1. Study Sites and Sampling
4.2. DNA Extraction
4.3. Bacterial Diversity: 16S rRNA Gene Amplicon Sequencing
4.4. Taxonomic Composition and Identification of Potentially Toxic Genus/Species
4.5. Graphical Representation of the Microbial Communities’ Structure Detected in the Reservoirs
4.6. Metagenomic Sequencing and Assembly of the Microcystis cf. aeruginosa CV01 Genome
4.7. Genome Annotation and General Features
4.8. Detection of Toxin Genes in M. cf. aeruginosa CV01
4.9. M. cf. aeruginosa CV01 Phylogenetic Analysis
4.10. Nucleotide Sequence Accession Numbers
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lobo de Pina, A. Hidroquímica e Qualidade das águas Subterrâneas da Ilha de Santiago—Cabo Verde. Ph.D. Thesis, Aveiro University, Aveiro, Portugal, 2009. [Google Scholar]
- Dodds, W.K.; Bouska, W.W.; Eitzmann, J.L.; Pilger, T.J.; Pitts, K.L.; Riley, A.J.; Schloesser, J.T.; Thornbrugh, D.J. Eutrophication of U.S. freshwaters: Analysis of potential economic damages. Environ. Sci. Technol. 2009, 43, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Heisler, J.; Glibert, P.M.; Burkholder, J.M.; Anderson, D.M.; Cochlan, W.; Dennison, W.C.; Dortch, Q.; Gobler, C.J.; Heil, C.A.; Humphries, E.; et al. Eutrophication and harmful algal blooms: A scientific consensus. Harmful Algae 2008, 8, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Paerl, H.W.; Paul, V.J. Climate change: Links to global expansion of harmful cyanobacteria. Water Res. 2012, 46, 1349–1363. [Google Scholar] [CrossRef] [PubMed]
- Paerl, H.W.; Scott, J.T. Throwing fuel on the fire: Synergistic effects of excessive nitrogen inputs and global warming on harmful algal blooms. Environ. Sci. Technol. 2010, 44, 7756–7758. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, J.M.; Davis, T.W.; Burford, M.A.; Gobler, C.J. The rise of harmful cyanobacteria blooms: The potential roles of eutrophication and climate change. Harmful Algae 2012, 14, 313–334. [Google Scholar] [CrossRef]
- Jones, S.E.; Newton, R.J.; McMahon, K.D. Evidence for structuring of bacterial community composition by organic carbon source in temperate lakes. Environ. Microbiol. 2009, 11, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.J.; Jones, S.E.; Eiler, A.; McMahon, K.D.; Bertilsson, S. A Guide to the Natural History of Freshwater Lake Bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 14–49. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Caro-Quintero, A.; Tsementzi, D.; DeLeon-Rodriguez, N.; Luo, C.; Poretsky, R.; Konstantinidis, K.T. Metagenomic insights into the evolution, function, and complexity of the planktonic microbial community of Lake Lanier, a temperate freshwater ecosystem. Appl. Environ. Microbiol. 2011, 77, 6000–6011. [Google Scholar] [CrossRef] [PubMed]
- Eiler, A.; Drakare, S.; Bertilsson, S.; Pernthaler, J.; Peura, S.; Rofner, C.; Simek, K.; Yang, Y.; Znachor, P.; Lindström, E.S. Unveiling Distribution Patterns of Freshwater Phytoplankton by a Next Generation Sequencing Based Approach. PLoS ONE 2013, 8, e53516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Q.; Bi, Y.; Deng, Y.; He, Z.; Wu, L.; Van Nostrand, J.D.; Shi, Z.; Li, J.; Wang, X.; Hu, Z.; et al. Impacts of the Three Gorges Dam on microbial structure and potential function. Nat. Sci. Rep. 2015, 5, 8605. [Google Scholar] [CrossRef] [PubMed]
- Eiler, A.; Bertilsson, S. Flavobacteria blooms in four eutrophic lakes: Linking population dynamics of freshwater bacterioplankton to resource availability. Appl. Environ. Microbiol. 2007, 73, 3511–3518. [Google Scholar] [CrossRef] [PubMed]
- Kosten, S.; Huszar, V.L.M.; Bécares, E.; Costa, L.S.; van Donk, E.; Hansson, L.A.; Jeppesen, E.; Kruk, C.; Lacerot, G.; Mazzeo, N.; et al. Warmer climates boost cyanobacterial dominance in shallow lakes. Glob. Chang. Biol. 2012, 18, 118–126. [Google Scholar] [CrossRef]
- Eiler, A.; Bertilsson, S. Composition of freshwater bacterial communities associated with cyanobacterial blooms in four Swedish lakes. Environ. Microbiol. 2004, 6, 1228–1243. [Google Scholar] [CrossRef] [PubMed]
- Conradie, K.R.; Barnard, S. The dynamics of toxic Microcystis strains and microcystin production in two hypertrofic South African reservoirs. Harmful Algae 2012, 20, 1–10. [Google Scholar] [CrossRef]
- Davis, T.W.; Berry, D.L.; Boyer, G.L.; Gobler, C.J. The effects of temperature and nutrients on the growth and dynamics of toxic and non-toxic strains of Microcystis during cyanobacteria blooms. Harmful Algae 2009, 8, 715–725. [Google Scholar] [CrossRef]
- Chorus, I.; Falconer, I.R.; Salas, H.J.; Bartram, J. Health risks caused by freshwater cyanobacteria in recreational waters. J. Toxicol. Environ. Heal. Part B 2000, 3, 323–347. [Google Scholar] [CrossRef]
- Paerl, H.W.; Hall, N.S.; Calandrino, E.S. Controlling harmful cyanobacterial blooms in a world experiencing anthropogenic and climatic-induced change. Sci. Total Environ. 2011, 409, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Paerl, H.W.; Huisman, J. Blooms Like It Hot. Science 2008, 320, 57–58. [Google Scholar] [CrossRef] [PubMed]
- Alvarenga, D.O.; Fiore, M.F.; Varani, A.M. A metagenomic approach to cyanobacterial genomics. Front. Microbiol. 2017, 8, 809. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Yeves, P.J.; Zemskay, T.I.; Rosselli, R.; Coutinho, F.H.; Zakharenko, A.S.; Blinov, V.V.; Rodriguez-Valera, F. Genomes of novel microbial lineages assembled from the sub-ice waters of Lake Baikal. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Stevens, S.L.R.; Chan, L.-K.; Bertilsson, S.; Glavina del Rio, T.; Tringe, S.G.; Malmstrom, R.R.; McMahon, K.D. Ecophysiology of freshwater Verrucomicrobia inferred from Metagenome-Assembled Genomes. mSphere 2017, 2, e00277-17. [Google Scholar] [CrossRef] [PubMed]
- Pinto, F.; Tett, A.; Armanini, F.; Asnicar, F.; Boscaini, A.; Pasolli, E.; Zolfo, M.; Donati, C.; Salmaso, N.; Segata, N. Draft genome sequences of novel Pseudomonas, Flavobacterium, and Sediminibacterium strains from a freshwater ecosystem. Genome Announc. 2018, 6, e00009-18. [Google Scholar] [CrossRef] [PubMed]
- Mehrshad, M.; Amoozegar, M.A.; Ghai, R.; Shahzadeh Fazeli, S.A.; Rodriguez-Valera, F. Genome reconstruction from metagenomic data sets reveals novel microbes in the brackish waters of the Caspian Sea. Appl. Environ. Microbiol. 2016, 82, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Kohler, E.; Grundler, V.; Haussinger, D.; Kurmayer, R.; Gademann, K.; Pernthaler, J.; Blom, J.F. The toxicity and enzyme activity of a chlorine and sulfate containing aeruginosin isolated from a non-microcystin-producing Planktothrix strain. Harmful Algae 2014, 39, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiblier, C.; Wood, S.; Echenique Subiabre, I.; Heath, M.; Villeneuve, A.; Humbert, J.-F. A review of current knowledge on toxic benthic freshwater cyanobacteria—Ecology, toxin production and risk management. Water Res. 2013, 47, 5464–5479. [Google Scholar] [CrossRef]
- Rounge, T.B.; Rohrlack, T.; Nederbragt, A.J.; Kristensen, T.; Jakobsen, K.S. A genome-wide analysis of nonribosomal peptide synthetase gene clusters and their peptides in a Planktothrix rubescens strain. BMC Genom. 2009, 10, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuergers, N.; Wilde, A. Appendages of the cyanobacterial cell. Life 2015, 700–715. [Google Scholar] [CrossRef] [PubMed]
- Nakasugi, K.; Alexova, R.; Svenson, C.J.; Neilan, B.A. Functional analysis of PilT from the toxic Cyanobacterium Microcystis aeruginosa PCC 7806. J. Bacteriol. 2007, 189, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Sandrini, G.; Huisman, J.; Matthijs, H.C.P. Potassium sensitivity differs among strains of the harmful cyanobacterium Microcystis and correlates with the presence of salt tolerance genes. FEMS Microbiol. Lett. 2015, 362, fnv121. [Google Scholar] [CrossRef] [PubMed]
- Sharon, I.; Alperovitch, A.; Rohwer, F.; Haynes, M.; Glaser, F.; Atamna-Ismaeel, N.; Pinter, R.Y.; Partensky, F.; Koonin, E.V; Wolf, Y.I.; et al. Photosystem I gene cassettes are present in marine virus genomes. Nature 2009, 461, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.; Gao, X.C.; Li, S.H.; Zhang, Q.Y. Genome analysis and gene nblA identification of Microcystis aeruginosa myovirus (MaMV-DC) reveal the evidence for horizontal gene transfer events between cyanomyovirus and host. J. Gen. Virol. 2015, 96, 3681–3697. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Nagasaki, K.; Takashima, Y.; Shirai, Y.; Tomaru, Y.; Takao, Y.; Sakamoto, S.; Hiroishi, S.; Ogata, H. Ma-LMM01 infecting toxic Microcystis aeruginosa illuminates diverse cyanophage genome strategies. J. Bacteriol. 2008, 190, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wang, K.; Jiao, N.; Chen, F. Genome sequences of siphoviruses infecting marine Synechococcus unveil a diverse cyanophage group and extensive phage–host genetic exchanges. Environ. Microbiol. 2012, 14, 540–558. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Axen, S.D.; Kerfeld, C.A. Evidence for the widespread distribution of CRISPR-Cas system in the Phylum Cyanobacteria. RNA Biol. 2013, 10, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Lin, F.; Li, Q.; Li, T.; Zhao, J. Comparative genomics reveals diversified CRISPR-Cas systems of globally distributed Microcystis aeruginosa, a freshwater bloom-forming cyanobacterium. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Cadel-Six, S.; Dauga, C.; Castets, A.M.; Rippka, R.; Bouchier, C.; Tandeau de Marsac, N.; Welker, M. Halogenase genes in nonribosomal peptide synthetase gene clusters of Microcystis (Cyanobacteria): Sporadic distribution and evolution. Mol. Biol. Evol. 2008, 25, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, T.; Ueda, A.; Nakano, T.; Nishizawa, A.; Miura, T.; Asayama, M.; Fujii, K.; Harada, K.; Shirai, M. Characterization of the locus of genes encoding enzymes producing heptadepsipeptide micropeptin in the unicellular cyanobacterium Microcystis. J. Biochem. 2011, 149, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, G.; Molitor, C.; Philmus, B.; Kurmayer, R. Nontoxic strains of cyanobacteria are the result of major gene deletion events induced by a transposable element. Mol. Biol. Evol. 2008, 25, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Humbert, J.-F.; Dorigo, U.; Cecchi, P.; Le Berre, B.; Debroas, D.; Bouvy, M. Comparison of the structure and composition of bacterial communities from temperate and tropical freshwater ecosystems. Environ. Microbiol. 2009, 11, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- McLellan, S.L.; Fisher, J.C.; Newton, R.J. The microbiome of urban waters. Int. Microbiol. 2015, 18, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Lindell, D.; Jaffe, J.D.; Johnson, Z.I.; Church, G.M.; Chisholm, S.W. Photosynthesis genes in marine viruses yield proteins during host infection. Nature 2005, 438, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Yoshida-Takashima, Y.; Yoshida, M.; Ogata, H.; Nagasaki, K.; Hiroishi, S.; Yoshida, T. Cyanophage infection in the bloom-forming cyanobacteria Microcystis aeruginosa in surface freshwater. Microbes Environ. 2012, 27, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Okello, W.; Portmann, C.; Erhard, M.; Gademann, K.; Kurmayer, R. Occurrence of microcystin-producing cyanobacteria in Ugandan freshwater habitats. Environ. Toxicol. 2010, 25, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Thomazeau, S.; Houdan-Fourmont, A.; Couté, A.; Duval, C.; Couloux, A.; Rousseau, F.; Bernard, C. The contribution of Sub-Saharan African strains to the phylogeny of cyanobacteria: Focusing on the nostocaceae (nostocales, cyanobacteria). J. Phycol. 2010, 46, 564–579. [Google Scholar] [CrossRef]
- Haande, S.; Rohrlack, T.; Ballot, A.; Røberg, K.; Skulberg, R.; Beck, M.; Wiedner, C. Genetic characterisation of Cylindrospermopsis raciborskii (Nostocales, Cyanobacteria) isolates from Africa and Europe. Harmful Algae 2008, 7, 692–701. [Google Scholar] [CrossRef]
- Berger, C.; Ba, N.; Gugger, M.; Bouvy, M.; Rusconi, F.; Couté, A.; Troussellier, M.; Bernard, C. Seasonal dynamics and toxicity of Cylindrospermopsis raciborskii in Lake Guiers (Senegal, West Africa). FEMS Microbiol. Ecol. 2006, 57, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Douma, M.; Ouahid, Y.; Campo, F.F.; Del Loudiki, M.; Mouhri, K.; Oudra, B. Identification and quantification of cyanobacterial toxins (microcystins) in two Moroccan drinking-water reservoirs (Mansour Eddahbi, Almassira). Environ. Monit. Assess. 2010, 160, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Matthews, M.; Bernard, S. Eutrophication and cyanobacteria in South Africa’s standing water bodies: A view from space. S. Afr. J. Sci. 2015, 111, 1–8. [Google Scholar] [CrossRef]
- Haande, S.; Ballot, A.; Rohrlack, T.; Fastner, J.; Wiedner, C.; Edvardsen, B. Diversity of Microcystis aeruginosa isolates (Chroococcales, Cyanobacteria) from East-African water bodies. Arch. Microbiol. 2007, 188, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ndlela, L.L.; Oberholster, P.J.; Van Wyk, J.H.; Cheng, P.H. An overview of cyanobacterial bloom occurrences and research in Africa over the last decade. Harmful Algae 2016, 60, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Englund, E.; Pattanaik, B.; Ubhayasekera, S.J.K.; Stensjö, K.; Bergquist, J.; Lindberg, P. Production of Squalene in Synechocystis sp. PCC 6803. PLoS ONE 2014, 9, e90270. [Google Scholar] [CrossRef] [PubMed]
- Zanchett, G.; Oliveira-Filho, E. Cyanobacteria and Cyanotoxins: From Impacts on Aquatic Ecosystems and Human Health to Anticarcinogenic Effects. Toxins (Basel) 2013, 5, 1896–1917. [Google Scholar] [CrossRef] [PubMed]
- Enzing, C.; Ploeg, M.; Barbosa, M.; Lolke, S. Microalgae-Based Products for the Food and Feed Sector: An Outlook for Europe; European Union: Luxembourg, 2014. [Google Scholar]
- Simas-Rodrigues, C.; Villela, H.D.M.; Martins, A.P.; Marques, L.G.; Colepicolo, P.; Tonon, A.P. Microalgae for economic applications: Advantages and perspectives for bioethanol. J. Exp. Bot. 2015, 66, 4097–4108. [Google Scholar] [CrossRef] [PubMed]
- Brennan, L.; Owende, P. Biofuels from microalgae—A review of technologies for production, processing, and extractions of biofuels and co-products. Renew. Sustain. Energy Rev. 2010, 14, 557–577. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high- throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.K.; Caruso, T.; Fischer, M.; Hancock, C.; Obermaier, E.; Prati, D.; Maier, T.S.; Meiners, T.; Caroline, M.; Wubet, T.; et al. Choosing and using diversity indices: Insights for ecological applications from the German Biodiversity Exploratories. Ecol. Evol. 2014, 4, 3514–3524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible rnteractive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Racine, J.S. RStudio: A platform-independent IDE for R and Sweave. J. Appl. Econom. 2012, 27, 167–172. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Humbert, J.-F.; Barbe, V.; Latifi, A.; Gugger, M.; Calteau, A.; Coursin, T.; Lajus, A.; Castelli, V.; Oztas, S.; Samson, G.; et al. A tribute to disorder in the genome of the bloom-forming freshwater cyanobacterium Microcystis aeruginosa. PLoS ONE 2013, 8, e70747. [Google Scholar] [CrossRef] [PubMed]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Goetz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. EGGNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR Recognition Tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Kall, L.; Krogh, A.; Sonnhammer, E.L.L. Advantages of combined transmembrane topology and signal peptide prediction—The Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. AntiSMASH 3.0—A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 4,918,369 | 100.0 |
| DNA coding (bp) | 3,900,077 | 79.3 |
| DNA G+C (bp) | 2,080,470 | 42.3 |
| DNA scaffolds | 262 | - |
| Total genes | 5484 | 100.0 |
| Protein-coding genes | 5051 | 92.1 |
| RNA genes | 45 | 0.8 |
| Genes with function prediction | 4996 | 91.1 |
| Genes assigned to COGs | 4166 | 76.0 |
| Genes with Pfam domains | 3407 | 62.1 |
| Genes with signal peptides | 112 | 2.0 |
| Genes with transmembrane helices | 3443 | 62.8 |
| CRISPR repeats | 9 | - |
| Reservoir | Location Coordinates | Start of Impoundment | Theoretical Maximum Volume (m3) | At Full Capacity? |
|---|---|---|---|---|
| Poilão | 15°04′25.0″ N,23°33′25.8″ W | 2006 | 1,200,000 | yes |
| Saquinho | 15°08′11.3″ N, 23°42′27.7″ W | 2013 | 563,000 | no |
| Faveta | 15°05′54.5″ N, 23°37′24.1″ W | 2013 | 536,565 | yes |
| F. Gorda | 15°07′5.8″ N, 23°35′36.5″ W | 2014 | 1,455,272 | empty |
| Salineiro | 14°57′03.5″ N, 23°38′00.4″ W | 2013 | 561,464 | empty |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semedo-Aguiar, A.P.; Pereira-Leal, J.B.; Leite, R.B. Microbial Diversity and Toxin Risk in Tropical Freshwater Reservoirs of Cape Verde. Toxins 2018, 10, 186. https://doi.org/10.3390/toxins10050186
Semedo-Aguiar AP, Pereira-Leal JB, Leite RB. Microbial Diversity and Toxin Risk in Tropical Freshwater Reservoirs of Cape Verde. Toxins. 2018; 10(5):186. https://doi.org/10.3390/toxins10050186
Chicago/Turabian StyleSemedo-Aguiar, Ana P., Jose B. Pereira-Leal, and Ricardo B. Leite. 2018. "Microbial Diversity and Toxin Risk in Tropical Freshwater Reservoirs of Cape Verde" Toxins 10, no. 5: 186. https://doi.org/10.3390/toxins10050186
APA StyleSemedo-Aguiar, A. P., Pereira-Leal, J. B., & Leite, R. B. (2018). Microbial Diversity and Toxin Risk in Tropical Freshwater Reservoirs of Cape Verde. Toxins, 10(5), 186. https://doi.org/10.3390/toxins10050186