Multicenter Evaluation of Independent High-Throughput and RT-qPCR Technologies for the Development of Analytical Workflows for Circulating miRNA Analysis


 , , , and
, , , and
Abstract
:1. Introduction
2. Results
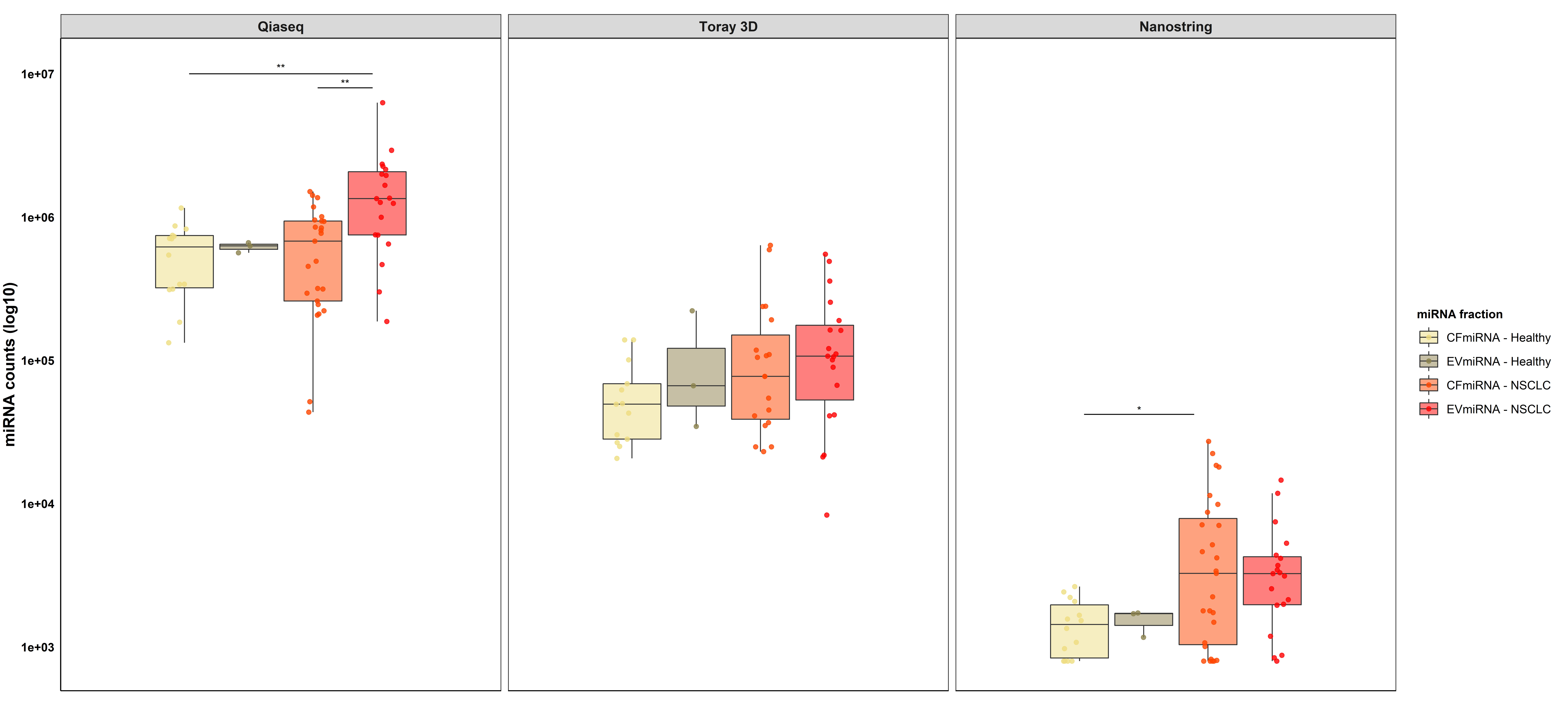
2.1. Screening Study
2.1.1. QIAseq Showed Higher Read Counts in Healthy Individuals and NSCLC Patients Compared to Hybridization Platforms
2.1.2. Systematic Comparisons Revealed Little Agreement between High-Throughput Platforms
2.2. Validation Study
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Plasma Preparation
4.3. Isolation of cfmiRNA and Extracellular Vesicle Derived miRNA (EVmiRNA)
4.4. Small RNA Sequencing Platforms
QIAseq miRNA Library Kit
4.5. Hybridization Platforms
4.5.1. NanoString nCounter Assay
4.5.2. Toray 3D
4.6. Quantitative PCR Platforms
4.6.1. miRCURY
4.6.2. Two-Tailed qPCR
4.7. Data Normalization
4.8. miRNA Candidate Selection
- (i)
- Control miRNAs (i.e. hemolysis controls miR-451a and miR-23a-3p, as well as negative controls widely found in urine samples (miR-30c-5p) and cerebrospinal fluid (CSF) (miR-124-3p)).
- (ii)
- miRNAs relevant for NSCLC diagnosis (i.e. miR-16-5p, miR-20a-5p, miR-21-5p, miR-125b-5p).
- (iii)
- Highly expressed miRNAs detected with QIAseq and Toray 3D.
- (iv)
- miRNAs which were statistically significant, differentially expressed in healthy control vs. patient samples (at least with one method).
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Babayan, A.; Pantel, K. Advances in liquid biopsy approaches for early detection and monitoring of cancer. Genome Med. 2018, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.H.D.; Bender, S.; Krahn, T.; Schlange, T. Ctdna and ctcs in liquid biopsy—Current status and where we need to progress. Comput. Struct. Biotechnol. J. 2018, 16, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Bullock, M.D.; Silva, A.M.; Kanlikilicer-Unaldi, P.; Filant, J.; Rashed, M.H.; Sood, A.K.; Lopez-Berestein, G.; Calin, G.A. Exosomal non-coding rnas: Diagnostic, prognostic and therapeutic applications in cancer. Non-Coding RNA 2015, 1, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.P.; Diallo, A.; Cruceanu, C.; Fiori, L.M.; Laboissiere, S.; Guillet, I.; Fontaine, J.; Ragoussis, J.; Benes, V.; Turecki, G.; et al. Biomarker discovery: Quantification of micrornas and other small non-coding rnas using next generation sequencing. BMC Med Genom. 2015, 8, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragomir, M.; Chen, B.; Calin, G.A. Exosomal lncrnas as new players in cell-to-cell communication. Transl. Cancer Res. 2018, 7, S243–S252. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chang, H.Y. Physiological roles of long noncoding rnas: Insight from knockout mice. Trends Cell Biol. 2014, 24, 594–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, K.V.; Mattick, J.S. The rise of regulatory rna. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayraktar, R.; Van Roosbroeck, K.; Calin, G.A. Cell-to-cell communication: Micrornas as hormones. Mol. Oncol. 2017, 11, 1673–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Liang, H.; Zhang, J.; Zen, K.; Zhang, C.Y. Secreted micrornas: A new form of intercellular communication. Trends Cell Biol. 2012, 22, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mrnas and micrornas is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating micrornas independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloten, V.; Neumann, M.H.D.; Di Pasquale, F.; Sprenger-Haussels, M.; Shaffer, J.M.; Schlumpberger, M.; Herdean, A.; Betsou, F.; Ammerlaan, W.; Af Hallstrom, T.; et al. Multicenter evaluation of circulating plasma microrna extraction technologies for the development of clinically feasible reverse transcription quantitative pcr and next-generation sequencing analytical work flows. Clin. Chem. 2019, 65, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Lampignano, R.; Kloten, V.; Krahn, T.; Schlange, T. Integrating circulating mirna analysis in the clinical management of lung cancer: Present or future? Mol. Asp. Med. 2020, 100844. [Google Scholar] [CrossRef] [PubMed]
- Giraldez, M.D.; Spengler, R.M.; Etheridge, A.; Godoy, P.M.; Barczak, A.J.; Srinivasan, S.; De Hoff, P.L.; Tanriverdi, K.; Courtright, A.; Lu, S.; et al. Comprehensive multi-center assessment of small rna-seq methods for quantitative mirna profiling. Nat. Biotechnol. 2018, 36, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.M.; Barczak, A.J.; DeHoff, P.; Srinivasan, S.; Etheridge, A.; Galas, D.; Das, S.; Erle, D.J.; Laurent, L.C. Comparison of reproducibility, accuracy, sensitivity, and specificity of mirna quantification platforms. Cell Rep. 2019, 29, 4212–4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestdagh, P.; Hartmann, N.; Baeriswyl, L.; Andreasen, D.; Bernard, N.; Chen, C.; Cheo, D.; D’Andrade, P.; DeMayo, M.; Dennis, L.; et al. Evaluation of quantitative mirna expression platforms in the microrna quality control (mirqc) study. Nat. Methods 2014, 11, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Androvic, P.; Valihrach, L.; Elling, J.; Sjoback, R.; Kubista, M. Two-tailed rt-qpcr: A novel method for highly accurate mirna quantification. Nucleic Acids Res. 2017, 45, e144. [Google Scholar] [CrossRef] [PubMed]
- Androvic, P.; Romanyuk, N.; Urdzikova-Machova, L.; Rohlova, E.; Kubista, M.; Valihrach, L. Two-tailed rt-qpcr panel for quality control of circulating microrna studies. Sci. Rep. 2019, 9, 4255. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| miRNA | EVmiRNA | cfmiRNA | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QIAseq | nCounter | Toray 3D | miRCURY | 2-Tailed qPCR | QIAseq | nCounter | Toray 3D | miRCURY | 2-Tailed qPCR | ||
| Screening Study | Validation Study | Screening Study | Validation Study | miRNA Class | |||||||
| miR-23a-3p | 0.076 | 0.059 | 0.398 | 0.000 n | 0.012 n | 0.120 | 0.306 | - | 0.317 | 0.001 n | Hemolysis controls |
| miR-451a | - | 0.363 | 0.297 | 0.002 n | 0.005 n | - | 0.072 | 0.235 | 0.001 n | 0.001 n | |
| miR-30c-5p | - | - | 0.004 H | 0.013 n | n* | 0.028 n | - | - | 0.174 | - | Negative controls |
| miR-124-3p | - | - | - | - | n* | - | - | - | 0.419 | 0.348 | |
| miR-16-5p | - | 0.386 | - | 0.000 n | 0.001 n | 0.000 n | 0.498 | - | 0.004 n | 0.004 n | Potentially relevant for NSCLC diagnosis |
| miR-20a-5p | - | - | - | 0.000 n | - | 0.220 | - | - | 0.135 | - | |
| miR-21-5p | - | 0.073 | - | 0.199 | 0.007 n | 0.000 n | - | - | 0.203 | 0.001 n | |
| miR-125b-5p | - | - | - | 0.004 n | n* | 0.138 | - | - | 0.184 | 0.067 | |
| let-7i-5p | - | - | - | 0.303 | 0.159 | 0.000 n | - | - | 0.166 | 0.003 n | Commonly found in "QIAseq cfmiRNA" and "miRCURY cfmiRNA" |
| miR-122-5p | - | - | - | 0.023 n | - | 0.005 n | 0.024 n | - | 0.185 | - | |
| miR-186-5p | - | 0.210 | - | 0.013 n | 0.005 H | 0.043 H | 0.477 | - | 0.033 n | 0.002 n | |
| miR-190b | 0.022 n | - | - | - | - | 0.000 n | - | - | 0.150 | - | |
| miR-191-5p | - | 0.030 n | - | 0.000 n | 0.004 n | 0.120 | - | - | 0.199 | - | |
| miR-199b-3p | - | 0.014 n | - | 0.021 n | - | 0.000 n | - | - | 0.227 | - | |
| miR-211-3p | - | - | 0.211 | n* | - | - | - | - | n* | - | |
| miR-365a-3p | - | 0.444 | 0.166 | 0.264 | 0.342 | - | - | - | 0.176 | 0.468 | |
| miR-144-3p | - | 0.047 H | - | 0.184 | - | 0.278 | 0.061 | - | 0.013 n | 0.131 | Commonly found in "QIAseq cfmiRNA" and "Toray cfmiRNA" |
| miR-671-5p | - | - | 0.360 | 0.001 n | n* | 0.194 | - | 0.345 | 0.077 | 0.444 | |
| miR-1909-3p | - | - | 0.446 | - | 0.131 | 0.000 H | - | 0.224 | 0.020 n | 0.292 | |
| miR-1914-5p | 0.006 n | - | - | - | 0.442 | 0.484 | - | - | 0.154 | 0.029 H | |
| miR-3195 | - | - | 0.500 | 0.019 n | n* | - | - | 0.336 | 0.098 | 0.366 | |
| miR-5739 | - | - | 0.403 | - | - | 0.012 H | - | 0.257 | - | n* | |
| miR-3185 | - | - | 0.058 | n* | 0.115 | 0.031 H | - | - | - | 0.488 | |
| miR-4433a | - | - | 0.289 | n* | n* | 0.352 | - | 0.353 | n* | 0.423 | |
| miR-5001-5p | 0.204 | - | 0.281 | - | n* | 0.130 | - | 0.050 n | - | 0.311 | |
| miR-6795-5p | - | - | 0.067 | - | - | - | - | 0.136 | - | - | |
| miRNAs detected, % | 15 | 35 | 50 | 62 | 42 | 81 | 23 | 31 | 77 | 65 | |
| Patients Characteristics | n | % |
|---|---|---|
| 27 | ||
| Median age (range) | 65 (52–82) | |
| Sex | ||
| Male | 17 | 63.0 |
| Female | 10 | 37.0 |
| Histology | ||
| Adeno | 25 | 92.6 |
| Squamos | 2 | 7.4 |
| Missing information | 0 | 0 |
| Tumor Size | ||
| T 1–2 | 8 | 29.6 |
| T 3–4 | 15 | 55.6 |
| Missing information | 4 | 14.8 |
| Lympnode Metastasis | ||
| n neg | 6 | 22.2 |
| n pos | 16 | 59.3 |
| Missing information | 5 | 18.5 |
| Distant Metastasis | ||
| M neg | 7 | 25.9 |
| M pos | 18 | 66.7 |
| Missing information | 2 | 7.4 |
| Smoking status | ||
| smoker | 10 | 37.0 |
| never smoker | 5 | 18.5 |
| former smoker | 3 | 11.1 |
| Missing information | 9 | 33.3 |
| Mutations | ||
| EGFR | 6 | 22.2 |
| KRAS | 7 | 25.9 |
| TP53 | 6 | 22.2 |
| Missing information or other mutations | 8 | 29.6 |
| Therapy (at blood draw) | ||
| Chemotherapy | 9 | 33.3 |
| Chemoradiotherapy | 3 | 11.1 |
| check point inhibitor | 10 | 37.0 |
| tyrosine kinase inhibitor | 5 | 18.5 |
| Therapy Response (at the next follow up) | ||
| Stable disease | 1 | 3.7 |
| Partial response | 0 | 0 |
| Complete response | 1 | 3.7 |
| Progression disease | 15 | 55.6 |
| Missing information | 10 | 37.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babayan, A.; Neumann, M.H.D.; Herdean, A.; Shaffer, J.M.; Janning, M.; Kobus, F.; Loges, S.; Di Pasquale, F.; Kubista, M.; Schlumpberger, M.; et al. Multicenter Evaluation of Independent High-Throughput and RT-qPCR Technologies for the Development of Analytical Workflows for Circulating miRNA Analysis. Cancers 2020, 12, 1166. https://doi.org/10.3390/cancers12051166
Babayan A, Neumann MHD, Herdean A, Shaffer JM, Janning M, Kobus F, Loges S, Di Pasquale F, Kubista M, Schlumpberger M, et al. Multicenter Evaluation of Independent High-Throughput and RT-qPCR Technologies for the Development of Analytical Workflows for Circulating miRNA Analysis. Cancers. 2020; 12(5):1166. https://doi.org/10.3390/cancers12051166
Chicago/Turabian StyleBabayan, Anna, Martin H. D. Neumann, Andrei Herdean, Jonathan M. Shaffer, Melanie Janning, Franca Kobus, Sonja Loges, Francesca Di Pasquale, Mikael Kubista, Martin Schlumpberger, and et al. 2020. "Multicenter Evaluation of Independent High-Throughput and RT-qPCR Technologies for the Development of Analytical Workflows for Circulating miRNA Analysis" Cancers 12, no. 5: 1166. https://doi.org/10.3390/cancers12051166
APA StyleBabayan, A., Neumann, M. H. D., Herdean, A., Shaffer, J. M., Janning, M., Kobus, F., Loges, S., Di Pasquale, F., Kubista, M., Schlumpberger, M., Lampignano, R., Krahn, T., Schlange, T., Sprenger-Haussels, M., Pantel, K., & Kloten, V., for the CANCER-ID consortium. (2020). Multicenter Evaluation of Independent High-Throughput and RT-qPCR Technologies for the Development of Analytical Workflows for Circulating miRNA Analysis. Cancers, 12(5), 1166. https://doi.org/10.3390/cancers12051166