
PSMA-Targeting Radiopharmaceuticals for Prostate Cancer Therapy: Recent Developments and Future Perspectives

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
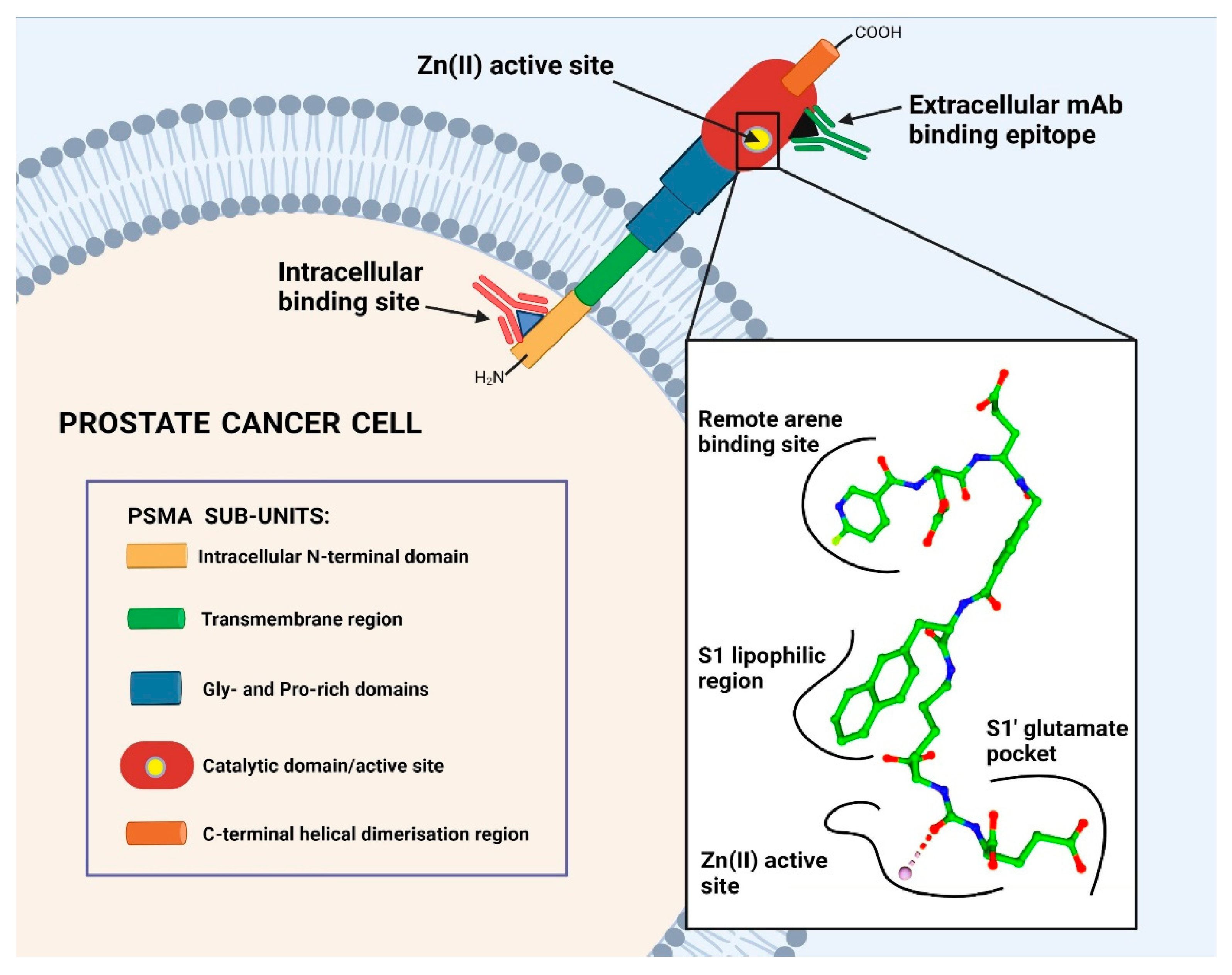
2. PSMA-Targeting Molecules: From mAbs to LMW Peptidomimetics
2.1. Monoclonal Antibodies (mAbs) Targeting PSMA
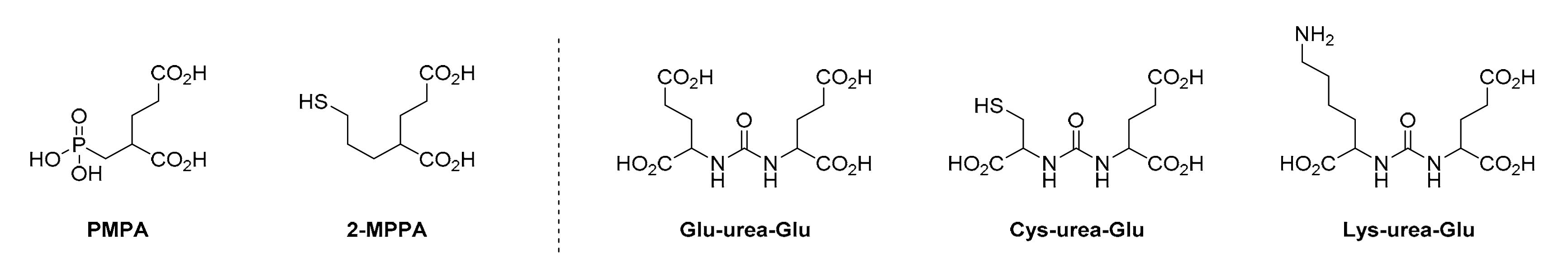
2.2. Low-Molecular Weight (LMW) PSMA Inhibitors
2.2.1. Binding Entity
2.2.2. Linkers and Chelators
2.2.3. Pharmacokinetic Modifications: Albumin Binders, Charged Spacers and Cleavable Linkers
2.3. Biomolecules vs. LMW-Inhibitors
3. Targeted Radionuclide Therapy of PSMA-Positive Prostate Cancer
3.1. Beta Minus (β-)-Based Targeted Radionuclide Therapy
3.1.1. β-Emitting Radionuclides for TRT
3.1.2. Preclinical PSMA β--TRT
3.1.3. Clinical PSMA β--TRT
3.2. Alpha (α)-Based Targeted Radionuclide Therapy
3.2.1. α-Emitting Radionuclides for TRT
3.2.2. Preclinical PSMA TAT
3.2.3. Clinical PSMA TAT
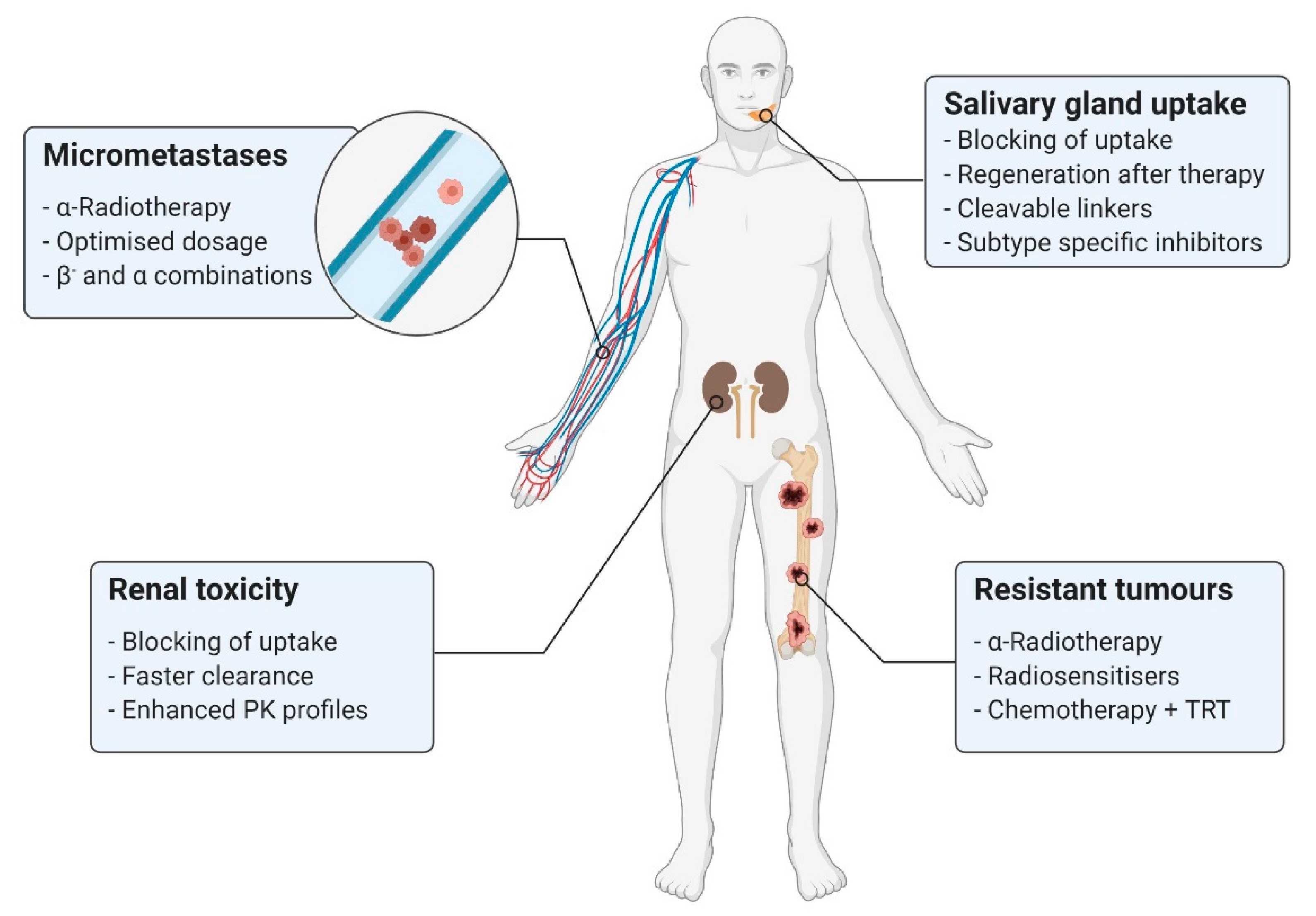
3.3. Enhancement of PSMA-Targeted Radionuclide Therapy
3.3.1. Combinatorial PSMA-Targeted Therapy
3.3.2. Side Effect Minimisation
4. Conclusions and Future Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Cancer Today. Available online: https://gco.iarc.fr/today/home (accessed on 18 May 2021).
- World Health Organization. Cancer Tomorrow. Available online: https://gco.iarc.fr/tomorrow/home (accessed on 18 May 2021).
- Hsing, A.W.; Tsao, L.; Devesa, S.S. International trends and patterns of prostate cancer incidence and mortality. Int. J. Cancer 2000, 85, 60–67. [Google Scholar] [CrossRef]
- He, L.; Fang, H.; Chen, C.; Wu, Y.; Wang, Y.; Ge, H.; Wang, L.; Wan, Y.; He, H. Metastatic castration-resistant prostate cancer: Academic insights and perspectives through bibliometric analysis. Medicine 2020, 99. [Google Scholar] [CrossRef] [PubMed]
- Mehtälä, J.; Zong, J.; Vassilev, Z.; Brobert, G.; Gabarró, M.S.; Stattin, P.; Khanfir, H. Overall survival and second primary malignancies in men with metastatic prostate cancer. PLoS ONE 2020, 15, e0227552. [Google Scholar] [CrossRef] [Green Version]
- Czerwińska, M.; Bilewicz, A.; Kruszewski, M.; Wegierek-Ciuk, A.; Lankoff, A. Targeted radionuclide therapy of prostate cancer-from basic research to clinical perspectives. Molecules 2020, 25, 1743. [Google Scholar] [CrossRef] [Green Version]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, S.S.; Reineke, T.M. Theranostics: Combining imaging and therapy. Bioconjug. Chem. 2011, 22, 1879–1903. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Oromieh, A.; Hetzheim, H.; Kratochwil, C.; Benesova, M.; Eder, M.; Neels, O.C.; Eisenhut, M.; Kübler, W.; Holland-Letz, T.; Giesel, F.L.; et al. The theranostic PSMA ligand PSMA-617 in the diagnosis of prostate cancer by PET/CT: Biodistribution in humans, radiation dosimetry, and first evaluation of tumor lesions. J. Nucl. Med. 2015, 56, 1697–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, C.; Domnanich, K.A.; Umbricht, C.A.; Van Der Meulen, N.P. Scandium and terbium radionuclides for radiotheranostics: Current state of development towards clinical application. Br. J. Radiol. 2018, 91. [Google Scholar] [CrossRef] [PubMed]
- Umbricht, C.A.; Benešová, M.; Schmid, R.M.; Türler, A.; Schibli, R.; van der Meulen, N.P.; Müller, C. 44Sc-PSMA-617 for radiotheragnostics in tandem with 177Lu-PSMA-617—preclinical investigations in comparison with 68Ga-PSMA-11 and 68Ga-PSMA-617. EJNMMI Res. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.W.; Cordon-Cardo2, C. Prostate-specific Membrane Antigen Expression in Normal and Malignant Human Tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar]
- O’Keefe, D.S.; Bacich, D.J.; Huang, S.S.; Heston, W.D.W. A perspective on the evolving story of PSMA biology, PSMA-based imaging, and endoradiotherapeutic strategies. J. Nucl. Med. 2018, 59, 1007–1013. [Google Scholar] [CrossRef]
- Davis, M.I.; Bennett, M.J.; Thomas, L.M.; Bjorkman, P.J. Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc. Natl. Acad. Sci. USA 2005, 102, 5981–5986. [Google Scholar] [CrossRef] [Green Version]
- Matthias, J.; Engelhardt, J.; Schäfer, M.; Bauder-Wüst, U.; Meyer, P.T.; Haberkorn, U.; Eder, M.; Kopka, K.; Hell, S.W.; Eder, A.C. Cytoplasmic localization of prostate-specific membrane antigen inhibitors may confer advantages for targeted cancer therapies. Cancer Res. 2021, 81, 2234–2245. [Google Scholar] [CrossRef]
- Horoszewicz, J.S.; Kawinski, E.; Murphy, G.P. Monoclonal antibodies to a new antigenic marker in epithelial prostatic cells and serum of prostatic cancer patients. Anticancer Res. 1987, 7, 927–935. [Google Scholar] [CrossRef]
- Lopes, A.D.; Davis, W.L.; Rosenstraus, M.J.; Uveges, A.J.; Gilman, S.C. Immunohistochemical and Pharmacokinetic Characterization of the Site-specific Immunoconjugate CYT-356 Derived from Antiprostate Monoclonal Antibody 7E11-C5. Cancer Res. 1990, 50, 6423–6429. [Google Scholar] [PubMed]
- Babaian, R.J.; Sayer, J.; Podoloff, D.A.; Steelhammer, L.C.; Bhadkamkar, V.A.; Gulfo, J.V. Radioimmunoscintigraphy of pelvic lymph nodes with 111indium-labeled monoclonal antibody CYT-356. J. Urol. 1994, 152, 1952–1955. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Akhtar, N.H.; Pail, O.; Saran, A.; Tyrell, L. Prostate-specific membrane antigen-based therapeutics. Adv. Urol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Smith-Jones, P.M.; Vallabahajosula, S.; Goldsmith, S.J.; Navarro, V.; Hunter, C.J.; Bastidas, D.; Bander, N.H. In vitro characterization of radiolabeled monoclonal antibodies specific for the extracellular domain of prostate-specific membrane antigen. Cancer Res. 2000, 60, 5237–5243. [Google Scholar] [PubMed]
- Liu, H.; Moy, P.; Kim, S.; Xia, Y.; Rajasekaran, A.; Navarro, V.; Knudsen, B.; Bander, N.H. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997, 57, 3629–3634. [Google Scholar] [PubMed]
- Smith-Jones, P.M.; Vallabhajosula, S.; Navarro, V.; Bastidas, D.; Goldsmith, S.J.; Bander, N.H. Radiolabeled monoclonal antibodies specific to the extracellular domain of prostate-specific membrane antigen: Preclinical studies in nude mice bearing LNCaP human prostate tumor. J. Nucl. Med. 2003, 44, 610–617. [Google Scholar] [CrossRef]
- Nanus, D.M.; Milowsky, M.I.; Kostakoglu, L.; Smith-Jones, P.M.; Vallabahajosula, S.; Goldsmith, S.J.; Bander, N.H.; Nelson, J.B.; Sellers, W.R.; Roach, M.; et al. Clinical use of monoclonal antibody huJ591 therapy: Targeting prostate specific membrane antigen. J. Urol. 2003, 170. [Google Scholar] [CrossRef]
- Pandit-Taskar, N.; O’Donoghue, J.A.; Durack, J.C.; Lyashchenko, S.K.; Cheal, S.M.; Beylergil, V.; Lefkowitz, R.A.; Carrasquillo, J.A.; Martinez, D.F.; Fung, A.M.; et al. A Phase I/II Study for Analytic Validation of 89Zr-J591 ImmunoPET as a Molecular Imaging Agent for Metastatic Prostate Cancer. Clin. Cancer Res. 2015, 21, 5277–5285. [Google Scholar] [CrossRef] [Green Version]
- Bander, N.H.; Milowsky, M.I.; Nanus, D.M.; Kostakoglu, L.; Vallabhajosula, S.; Goldsmith, S.J. Phase I trial of 177Lutetium-labeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J. Clin. Oncol. 2005, 23, 4591–4601. [Google Scholar] [CrossRef]
- Regino, C.A.S.; Wong, K.J.; Milenic, D.E.; Holmes, E.H.; Garmestani, K.; Choyke, P.L.; Brechbiel, M.W. Preclinical evaluation of a monoclonal antibody (3C6) specific for prostate-specific membrane antigen. Curr. Radiopharm. 2009, 2, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alt, K.; Wiehr, S.; Ehrlichmann, W.; Reischl, G.; Wolf, P.; Pichler, B.J.; Elsässer-Beile, U.; Bühler, P. High-resolution animal PET imaging of prostate cancer xenografts with three different 64Cu-labeled antibodies against native cell-adherent PSMA. Prostate 2010, 70, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Colombatti, M.; Grasso, S.; Porzia, A.; Fracasso, G.; Scupoli, M.T.; Cingarlini, S.; Poffe, O.; Naim, H.Y.; Heine, M.; Tridente, G.; et al. The prostate specific membrane antigen regulates the expression of IL-6 and CCL5 in prostate tumour cells by activating the MAPK pathways. PLoS ONE 2009, 4, e4608. [Google Scholar] [CrossRef]
- Lütje, S.; Rijpkema, M.; Franssen, G.M.; Fracasso, G.; Helfrich, W.; Eek, A.; Oyen, W.J.; Colombatti, M.; Boerman, O.C. Dual-modality image-guided surgery of prostate cancer with a radiolabeled fluorescent anti-PSMA monoclonal antibody. J. Nucl. Med. 2014, 55, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, S.T.; Milowsky, M.I.; Morris, M.; Vallabhajosula, S.; Christos, P.; Akhtar, N.H.; Osborne, J.; Goldsmith, S.J.; Larson, S.; Taskar, N.P.; et al. Phase II study of lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 5182–5191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, M.S.; McGonigle, P.; Hornby, P.J. The pharmacology and therapeutic applications of monoclonal antibodies. Pharmacol. Res. Perspect. 2019, 7, e00535. [Google Scholar] [CrossRef] [PubMed]
- Viola-Villegas, N.T.; Sevak, K.K.; Carlin, S.D.; Doran, M.G.; Evans, H.W.; Bartlett, D.W.; Wu, A.M.; Lewis, J.S. Noninvasive imaging of PSMA in prostate tumors with 89Zr-Labeled huJ591 engineered antibody fragments: The faster alternatives. Mol. Pharm. 2014, 11, 3965–3973. [Google Scholar] [CrossRef] [Green Version]
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, E.Y.; Shah, K. Nanobodies: Next Generation of Cancer Diagnostics and Therapeutics. Front. Oncol. 2020, 10, 1182. [Google Scholar] [CrossRef]
- Evazalipour, M.; D’Huyvetter, M.; Tehrani, B.S.; Abolhassani, M.; Omidfar, K.; Abdoli, S.; Arezumand, R.; Morovvati, H.; Lahoutte, T.; Muyldermans, S.; et al. Generation and characterization of nanobodies targeting PSMA for molecular imaging of prostate cancer. Contrast Media Mol. Imaging 2014, 9, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Chatalic, K.L.S.; Veldhoven-Zweistra, J.; Bolkestein, M.; Hoeben, S.; Koning, G.A.; Boerman, O.C.; De Jong, M.; Van Weerden, W.M. A novel 111In-labeled anti-prostate-specific membrane antigen nanobody for targeted SPECT/CT imaging of prostate cancer. J. Nucl. Med. 2015, 56, 1094–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiffany, C.W.; Lapidus, R.G.; Merion, A.; Calvin, D.C.; Slusher, B.S. Characterization of the enzymatic activity of PSM: Comparison with brain NAALADase. Prostate 1999, 39, 28–35. [Google Scholar] [CrossRef]
- Luthi-Carter, R.; Barczak, A.K.; Speno, H.; Coyle, J.T. Molecular characterization of human brain N-acetylated α-linked acidic dipeptidase (NAALADase). J. Pharmacol. Exp. Ther. 1998, 286, 1020–1025. [Google Scholar]
- Heston, W. Bedeutung des prostataspezifischen Membranantigens (PSMA). Urologe 1996, 35, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.F.; Cole, D.C.; Slusher, B.S.; Stetz, S.L.; Ross, L.E.; Donzanti, B.A.; Trainor, D.A. Design, synthesis, and biological activity of a potent inhibitor of the neuropeptidase N-acetylated α-linked acidic dipeptidase. J. Med. Chem. 1996, 39, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.; Slusher, B. Design of NAALADase Inhibitors A Novel Neuroprotective Strategy. Curr. Med. Chem. 2001, 8, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Lesche, R.; Kettschau, G.; Gromov, A.V.; Böhnke, N.; Borkowski, S.; Mönning, U.; Hegele-Hartung, C.; Döhr, O.; Dinkelborg, L.M.; Graham, K. Preclinical evaluation of BAY 1075553, a novel 18F-labelled inhibitor of prostate-specific membrane antigen for PET imaging of prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 89–101. [Google Scholar] [CrossRef]
- Beheshti, M.; Kunit, T.; Haim, S.; Zakavi, R.; Schiller, C.; Stephens, A.; Dinkelborg, L.; Langsteger, W.; Loidl, W. BAY 1075553 PET-CT for Staging and Restaging Prostate Cancer Patients: Comparison with [18F] Fluorocholine PET-CT (Phase I Study). Mol. Imaging Biol. 2015, 17, 424–433. [Google Scholar] [CrossRef]
- Jackson, P.F.; Tays, K.L.; Maclin, K.M.; Ko, Y.S.; Li, W.; Vitharana, D.; Tsukamoto, T.; Stoermer, D.; Lu, X.C.M.; Wozniak, K.; et al. Design and pharmacological activity of phosphinic acid based NAALADase inhibitors. J. Med. Chem. 2001, 44, 4170–4175. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Ng, R.J.; Shieh, C.C.; Martinez, A.R.; Berkman, C.E. Inhibition of glutamate carboxypeptidase by phosphoryl and thiophosphoryl derivatives of glutamic and 2-hydroxyglutaric acid. Phosphorus Sulfur Silicon Relat. Elem. 2003, 178, 17–32. [Google Scholar] [CrossRef]
- Maung, J.; Mallari, J.P.; Girtsman, T.A.; Wu, L.Y.; Rowley, J.A.; Santiago, N.M.; Brunelle, A.N.; Berkman, C.E. Probing for a hydrophobic a binding register in prostate-specific membrane antigen with phenylalkylphosphonamidates. Bioorganic Med. Chem. 2004, 12, 4969–4979. [Google Scholar] [CrossRef] [PubMed]
- Lapi, S.E.; Wahnishe, H.; Pham, D.; Wu, L.Y.; Nedrow-Byers, J.R.; Liu, T.; Vejdani, K.; VanBrocklin, H.F.; Berkman, C.E.; Jones, E.F. Assessment of an 18F-labeled phosphoramidate peptidomimetic as a new prostate-specific membrane antigen-targeted imaging agent for prostate cancer. J. Nucl. Med. 2009, 50, 2042–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedrow-Byers, J.R.; Moore, A.L.; Ganguly, T.; Hopkins, M.R.; Fulton, M.D.; Benny, P.D.; Berkman, C.E. PSMA-targeted SPECT agents: Mode of binding effect on in vitro performance. Prostate 2013, 73, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Ley, C.R.; Beattie, N.R.; Dannoon, S.; Regan, M.; VanBrocklin, H.; Berkman, C.E. AccessSynthesis and Evaluation of Constrained Phosphoramidate Inhibitors of Prostate-Specific Membrane Antigen. Bioorganic Med. Chem. Lett. 2015, 25, 2536–2539. [Google Scholar] [CrossRef]
- Ganguly, T.; Dannoon, S.; Hopkins, M.R.; Murphy, S.; Cahaya, H.; Blecha, J.E.; Jivan, S.; Drake, C.R.; Barinka, C.; Jones, E.F.; et al. A high-affinity [18F]-labeled phosphoramidate peptidomimetic PSMA-targeted inhibitor for PET imaging of prostate cancer. Nucl. Med. Biol. 2015, 42, 780–787. [Google Scholar] [CrossRef] [Green Version]
- Dannoon, S.; Ganguly, T.; Cahaya, H.; Geruntho, J.J.; Galliher, M.S.; Beyer, S.K.; Choy, C.J.; Hopkins, M.R.; Regan, M.; Blecha, J.E.; et al. Structure-Activity Relationship of 18F-Labeled Phosphoramidate Peptidomimetic Prostate-Specific Membrane Antigen (PSMA)-Targeted Inhibitor Analogues for PET Imaging of Prostate Cancer. J. Med. Chem. 2016, 59, 5684–5694. [Google Scholar] [CrossRef]
- Choy, C.J.; Ling, X.; Geruntho, J.J.; Beyer, S.K.; Latoche, J.D.; Langton-Webster, B.; Anderson, C.J.; Berkman, C.E. 177Lu-labeled phosphoramidate-based PSMA inhibitors: The effect of an albumin binder on biodistribution and therapeutic efficacy in prostate tumor-bearing mice. Theranostics 2017, 7, 1928–1939. [Google Scholar] [CrossRef]
- Behr, S.C.; Aggarwal, R.; VanBrocklin, H.F.; Flavell, R.R.; Gao, K.; Small, E.J.; Blecha, J.; Jivan, S.; Hope, T.A.; Simko, J.P.; et al. Phase i study of CTT1057, an 18F-labeled imaging agent with phosphoramidate core targeting prostate-specific membrane antigen in prostate cancer. J. Nucl. Med. 2019, 60, 910–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majer, P.; Jackson, P.F.; Delahanty, G.; Grella, B.S.; Ko, Y.S.; Li, W.; Liu, Q.; Maclin, K.M.; Poláková, J.; Shaffer, K.A.; et al. Synthesis and biological evaluation of thiol-based inhibitors of glutamate carboxypeptidase II: Discovery of an orally active GCP II inhibitor. J. Med. Chem. 2003, 46, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Majer, P.; Hin, B.; Stoermer, D.; Adams, J.; Xu, W.; Duvall, B.R.; Delahanty, G.; Liu, Q.; Stathis, M.J.; Wozniak, K.M.; et al. Structural optimization of thiol-based inhibitors of glutamate carboxypeptidase II by modification of the P1′ side chain. J. Med. Chem. 2006, 49, 2876–2885. [Google Scholar] [CrossRef] [PubMed]
- Van Der Post, J.P.; De Visser, S.J.; De Kam, M.L.; Woelfler, M.; Hilt, D.C.; Vornov, J.; Burak, E.S.; Bortey, E.; Slusher, B.S.; Limsakun, T.; et al. The central nervous system effects, pharmacokinetics and safety of the NAALADase-inhibitor GPI 5693. Br. J. Clin. Pharmacol. 2005, 60, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Stoermer, D.; Vitharana, D.; Hin, N.; Delahanty, G.; Duvall, B.; Ferraris, D.V.; Grella, B.S.; Hoover, R.; Rojas, C.; Shanholtz, M.K.; et al. Design, synthesis, and pharmacological evaluation of glutamate carboxypeptidase II (GCPII) inhibitors based on thioalkylbenzoic acid scaffolds. J. Med. Chem. 2012, 55, 5922–5932. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, D.V.; Majer, P.; Ni, C.; Slusher, C.E.; Rais, R.; Wu, Y.; Wozniak, K.M.; Alt, J.; Rojas, C.; Slusher, B.S.; et al. δ-thiolactones as prodrugs of thiol-based glutamate carboxypeptidase II (GCPII) inhibitors. J. Med. Chem. 2014, 57, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, W.; Cai, H.; Chen, L.; Jin, X.; Liao, X.; Jia, Z. Recent Advances in Prostate-Specific Membrane Antigen-Based Radiopharmaceuticals. Curr. Top. Med. Chem. 2019, 19, 33–56. [Google Scholar] [CrossRef] [PubMed]
- Siow, A.; Kowalczyk, R.; Brimble, M.A.; Harris, P.W.R. Evolution of Peptide-Based Prostate-Specific Membrane Antigen (PSMA) Inhibitors: An Approach to Novel Prostate Cancer Therapeutics. Curr. Med Chem. 2021, 28, 3713–3752. [Google Scholar] [CrossRef]
- Ha, H.; Kwon, H.; Lim, T.; Jang, J.; Park, S.-K.; Byun, Y. Inhibitors of prostate-specific membrane antigen in the diagnosis and therapy of metastatic prostate cancer–a review of patent literature. Expert Opin. Ther. Pat. 2021, 31, 525–547. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Nan, F.; Conti, P.; Zhang, J.; Ramadan, E.; Bzdega, T.; Wroblewska, B.; Neale, J.H.; Pshenichkin, S.; Wroblewski, J.T. Design of remarkably simple, yet potent urea-based inhibitors of glutamate carboxypeptidase II (NAALADase). J. Med. Chem. 2001, 44, 298–301. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Zhang, J.; Nan, F.; Petukhov, P.A.; Grajkowska, E.; Wroblewski, J.T.; Yamamoto, T.; Bzdega, T.; Wroblewska, B.; Neale, J.H. Synthesis of Urea-Based Inhibitors as Active Site Probes of Glutamate Carboxypeptidase II: Efficacy as Analgesic Agents. J. Med. Chem. 2004, 47, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Felber, V.B.; Valentin, M.A.; Wester, H.-J. Design of PSMA ligands with modifications at the inhibitor part: An approach to reduce the salivary gland uptake of radiolabeled PSMA inhibitors? EJNMMI Radiopharm. Chem. 2021, 6, 1–24. [Google Scholar] [CrossRef]
- Foss, C.A.; Mease, R.C.; Fan, H.; Wang, Y.; Ravert, H.T.; Dannals, R.F.; Olszewski, R.T.; Heston, W.D.; Kozikowski, A.P.; Pomper, M.G. Radiolabeled small-molecule ligands for prostate-specific membrane antigen: In vivo imaging in experimental models of prostate cancer. Clin. Cancer Res. 2005, 11, 4022–4028. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Foss, C.; Mease, R.; Nimmagadda, S.; Fox, J.; Kozikowski, A.; Pomper, M. Synthesis, biodistribution, and experimental prostate tumor imaging of p-[125I]iodobenzoyl-lys-NH(CO)NH-glu. J. Nucl. Med. 2007, 48 (Suppl. 2), 19P. [Google Scholar]
- Barinka, C.; Byun, Y.; Dusich, C.L.; Banerjee, S.R.; Chen, Y.; Castanares, M.; Kozikowski, A.P.; Mease, R.C.; Pomper, M.G.; Lubkowski, J. Interactions between human glutamate carboxypeptidase II and urea-based inhibitors: Structural characterization. J. Med. Chem. 2008, 51, 7737–7743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Kwon, H.; Barinka, C.; Motlova, L.; Nam, S.; Choi, D.; Ha, H.; Nam, H.; Son, S.H.; Minn, I.; et al. Novel β- And γ-Amino Acid-Derived Inhibitors of Prostate-Specific Membrane Antigen. J. Med. Chem. 2020, 63, 3261–3273. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Lim, H.; Ha, H.; Choi, D.; Son, S.H.; Nam, H.; Minn, I.; Byun, Y. Structure-activity relationship studies of prostate-specific membrane antigen (PSMA) inhibitors derived from α-amino acid with (S)- or (R)-configuration at P1′ region. Bioorg. Chem. 2020, 104, 104304. [Google Scholar] [CrossRef] [PubMed]
- Choy, C.J.; Fulton, M.D.; Davis, A.L.; Hopkins, M.; Choi, J.K.; Anderson, M.O.; Berkman, C.E. Rationally designed sulfamides as glutamate carboxypeptidase II inhibitors. Chem. Biol. Drug Des. 2013, 82, 612–619. [Google Scholar] [CrossRef]
- Yang, X.; Mease, R.C.; Pullambhatla, M.; Lisok, A.; Chen, Y.; Foss, C.A.; Wang, Y.; Shallal, H.; Edelman, H.; Hoye, A.T.; et al. Fluorobenzoyllysinepentanedioic Acid Carbamates: New Scaffolds for Positron Emission Tomography (PET) Imaging of Prostate-Specific Membrane Antigen (PSMA). J. Med. Chem. 2016, 59, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Barinka, C.; Novakova, Z.; Hin, N.; Bím, D.; Ferraris, D.V.; Duvall, B.; Kabarriti, G.; Tsukamoto, R.; Budesinsky, M.; Motlova, L.; et al. Structural and computational basis for potent inhibition of glutamate carboxypeptidase II by carbamate-based inhibitors. Bioorganic Med. Chem. 2019, 27, 255–264. [Google Scholar] [CrossRef]
- Young, J.D.; Ma, M.T.; Eykyn, T.R.; Atkinson, R.A.; Abbate, V.; Cilibrizzi, A.; Hider, R.C.; Blower, P.J. Dipeptide Inhibitors of the Prostate Specific Membrane Antigen (PSMA): A Comparison of Urea and Thiourea Derivatives. Bioorg. Med. Chem. Lett. 2021, 128044. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Singh, P.; Topaloglu, O.; Isaacs, J.T.; Demneade, S.R. A dimeric peptide that binds selectively to prostate-specific membrane antigen and inhibits its enzymatic activity. Cancer Res. 2006, 66, 9171–9177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupold, S.E.; Rodriguez, R. Disulfide-constrained peptides that bind to the extracellular portion of the prostate-specific membrane antigen. Mol. Cancer Ther. 2004, 3, 597–603. [Google Scholar]
- Zhang, A.X.; Murelli, R.P.; Barinka, C.; Michel, J.; Cocleaza, A.; Jorgensen, W.L.; Lubkowski, J.; Spiegel, D.A. A Remote arene-binding site on prostate specific membrane antigen revealed by antibody-recruiting small molecules. J. Am. Chem. Soc. 2010, 132, 12711–12716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zechmann, C.M.; Afshar-Oromieh, A.; Armor, T.; Stubbs, J.B.; Mier, W.; Hadaschik, B.; Joyal, J.; Kopka, K.; Debus, J.; Babich, J.W.; et al. Radiation dosimetry and first therapy results with a 124I/131I-labeled small molecule (MIP-1095) targeting PSMA for prostate cancer therapy. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1280–1292. [Google Scholar] [CrossRef] [Green Version]
- Kiess, A.P.; Minn, I.; Vaidyanathan, G.; Hobbs, R.F.; Josefsson, A.; Shen, C.; Brummet, M.; Chen, Y.; Choi, J.; Koumarianou, E.; et al. (2S)-2-(3-(1-carboxy-5-(4-211At-astatobenzamido)pentyl) ureido)-pentanedioic acid for PSMA-targeted α-particle radiopharmaceutical therapy. J. Nucl. Med. 2016, 57, 1569–1575. [Google Scholar] [CrossRef] [Green Version]
- Barrett, J.A.; Coleman, R.E.; Goldsmith, S.J.; Vallabhajosula, S.; Petry, N.A.; Cho, S.; Armor, T.; Stubbs, J.B.; Maresca, K.P.; Stabin, M.G.; et al. First-in-man evaluation of 2 high-affinity PSMA-avid small molecules for imaging prostate cancer. J. Nucl. Med. 2013, 54, 380–387. [Google Scholar] [CrossRef] [Green Version]
- Hillier, S.M.; Maresca, K.P.; Femia, F.J.; Marquis, J.C.; Foss, C.A.; Nguyen, N.; Zimmerman, C.N.; Barrett, J.A.; Eckelman, W.C.; Pomper, M.G.; et al. Preclinical evaluation of novel glutamate-urea-lysine analogues that target prostate-specific membrane antigen as molecular imaging pharmaceuticals for prostate cancer. Cancer Res. 2009, 69, 6932–6940. [Google Scholar] [CrossRef] [Green Version]
- Childers, A.; Jeffrey, W.; Ii, W. PSMA Targeted Radiohaloge-Nated Ureas for Cancer Radiotherapy. WO2017070482A2, 27 April 2017. [Google Scholar]
- Vaidyanathan, G.; Mease, R.C.; Minn, I.; Choi, J.; Chen, Y.; Shallal, H.; Kang, C.M.; McDougald, D.; Kumar, V.; Pomper, M.G.; et al. Synthesis and preliminary evaluation of 211At-labeled inhibitors of prostate-specific membrane antigen for targeted alpha particle therapy of prostate cancer. Nucl. Med. Biol. 2021, 94–95, 67–80. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, Y.; Zeng, Q.; Xie, T.; Yao, S.; Zhang, J.; Cui, M. Synthesis, Preclinical Evaluation, and First-in-Human PET Study of Quinoline-Containing PSMA Tracers with Decreased Renal Excretion. J. Med. Chem. 2021, 64, 4179–4195. [Google Scholar] [CrossRef]
- Banerjee, S.R.; Pullambhatla, M.; Byun, Y.; Nimmagadda, S.; Green, G.; Fox, J.J.; Horti, A.; Mease, R.C.; Pomper, M.G. 68Ga-labeled inhibitors of prostate-specific membrane antigen (PSMA) for imaging prostate cancer. J. Med. Chem. 2010, 53, 5333–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eder, M.; Schäfer, M.; Bauder-Wüst, U.; Hull, W.E.; Wängler, C.; Mier, W.; Haberkorn, U.; Eisenhut, M. 68Ga-complex lipophilicity and the targeting property of a urea-based PSMA inhibitor for PET imaging. Bioconjug. Chem. 2012, 23, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Oromieh, A.; Malcher, A.; Eder, M.; Eisenhut, M.; Linhart, H.G.; Hadaschik, B.A.; Holland-Letz, T.; Giesel, F.L.; Kratochwil, C.; Haufe, S.; et al. Pet imaging with a [68Ga]gallium-labelled psma ligand for the diagnosis of prostate cancer: Biodistribution in humans and first evaluation of tumour lesions. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 486–495. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration; Center for Drug Evaluation and Research. Gallium Ga-68 PSMA-11 Injection Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/212643Orig1s000Approv.pdf (accessed on 15 June 2021).
- Huang, S.S.; Wang, X.; Zhang, Y.; Doke, A.; Difilippo, F.P.; Heston, W.D. Improving the biodistribution of PSMA-targeting tracers with a highly negatively charged linker. Prostate 2014, 74, 702–713. [Google Scholar] [CrossRef]
- Baranski, A.C.; Schäfer, M.; Bauder-Wüst, U.; Wacker, A.; Schmidt, J.; Liolios, C.; Mier, W.; Haberkorn, U.; Eisenhut, M.; Kopka, K.; et al. Improving the Imaging Contrast of 68Ga-PSMA-11 by Targeted Linker Design: Charged Spacer Moieties Enhance the Pharmacokinetic Properties. Bioconjug. Chem. 2017, 28, 2485–2492. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, J.; Roscher, M.; Schäfer, M.; Geerlings, M.; Benešová, M.; Bauder-Wüst, U.; Remde, Y.; Eder, M.; Nováková, Z.; Motlová, L.; et al. Development of PSMA-1007-Related Series of 18F-Labeled Glu-Ureido-Type PSMA Inhibitors. J. Med. Chem. 2020, 63, 10897–10907. [Google Scholar] [CrossRef]
- Cardinale, J.; Schäfer, M.; Benesova, M.; Bauder-Wüst, U.; Leotta, K.; Eder, M.; Neels, O.C.; Haberkorn, U.; Giesel, F.L.; Kopka, K. Preclinical evaluation of 18F-PSMA-1007, a new prostate-specific membrane antigen ligand for prostate cancer imaging. J. Nucl. Med. 2017, 58, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Benesová, M.; Schäfer, M.; Bauder-Wüst, U.; Afshar-Oromieh, A.; Kratochwil, C.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Preclinical evaluation of a tailor-made DOTA-conjugated PSMA inhibitor with optimized linker moiety for imaging and endoradiotherapy of prostate cancer. J. Nucl. Med. 2015, 56, 914–920. [Google Scholar] [CrossRef] [Green Version]
- Weineisen, M.; Schottelius, M.; Simecek, J.; Baum, R.P.; Yildiz, A.; Beykan, S.; Kulkarni, H.R.; Lassmann, M.; Klette, I.; Eiber, M.; et al. 68Ga- and 177Lu-labeled PSMA I and T: Optimization of a PSMA-targeted theranostic concept and first proof-of-concept human studies. J. Nucl. Med. 2015, 56, 1169–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weineisen, M.; Simecek, J.; Schottelius, M.; Schwaiger, M.; Wester, H.J. Synthesis and preclinical evaluation of DOTAGA-conjugated PSMA ligands for functional imaging and endoradiotherapy of prostate cancer. EJNMMI Res. 2014, 4, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Benešová, M.; Bauder-Wüst, U.; Schäfer, M.; Klika, K.D.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Linker Modification Strategies to Control the Prostate-Specific Membrane Antigen (PSMA)-Targeting and Pharmacokinetic Properties of DOTA-Conjugated PSMA Inhibitors. J. Med. Chem. 2016, 59, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.T.; Pan, J.; Zhang, Z.; Lau, J.; Merkens, H.; Zhang, C.; Colpo, N.; Lin, K.S.; Bénard, F. Effects of Linker Modification on Tumor-to-Kidney Contrast of 68Ga-Labeled PSMA-Targeted Imaging Probes. Mol. Pharm. 2018, 15, 3502–3511. [Google Scholar] [CrossRef] [PubMed]
- Wüstemann, T.; Bauder-Wüst, U.; Schäfer, M.; Eder, M.; Benesova, M.; Leotta, K.; Kratochwil, C.; Haberkorn, U.; Kopka, K.; Mier, W. Design of internalizing PSMA-specific glu-ureido-based radiotherapeuticals. Theranostics 2016, 6, 1085–1095. [Google Scholar] [CrossRef] [Green Version]
- Dennis, M.S.; Zhang, M.; Gloria Meng, Y.; Kadkhodayan, M.; Kirchhofer, D.; Combs, D.; Damico, L.A. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J. Biol. Chem. 2002, 277, 35035–35043. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.M.; Amor-Coarasa, A.; Nikolopoulou, A.; Wüstemann, T.; Barelli, P.; Kim, D.; Williams, C.; Zheng, X.; Bi, C.; Hu, B.; et al. Dual-target binding ligands with modulated pharmacokinetics for endoradiotherapy of prostate cancer. J. Nucl. Med. 2017, 58, 1442–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Jacobson, O.; Tian, R.; Mease, R.C.; Kiesewetter, D.O.; Niu, G.; Pomper, M.G.; Chen, X. Radioligand Therapy of Prostate Cancer with a Long-Lasting Prostate-Specific Membrane Antigen Targeting Agent 90Y-DOTA-EB-MCG. Bioconjug. Chem. 2018, 29, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tian, R.; Niu, G.; Ma, Y.; Lang, L.; Szajek, L.P.; Kiesewetter, D.O.; Jacobson, O.; Chen, X. Single low-dose injection of evans blue modified PSMA-617 radioligand therapy eliminates prostate-specific membrane antigen positive tumors. Bioconjug. Chem. 2018, 29, 3213–3221. [Google Scholar] [CrossRef]
- Benešová, M.; Umbricht, C.A.; Schibli, R.; Müller, C. Albumin-Binding PSMA Ligands: Optimization of the Tissue Distribution Profile. Mol. Pharm. 2018, 15, 934–946. [Google Scholar] [CrossRef]
- Umbricht, C.A.; Benešová, M.; Schibli, R.; Müller, C. Preclinical Development of Novel PSMA-Targeting Radioligands: Modulation of Albumin-Binding Properties to Improve Prostate Cancer Therapy. Mol. Pharm. 2018, 15, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.T.; Lin, K.S.; Zhang, Z.; Uribe, C.F.; Merkens, H.; Zhang, C.; Bénard, F. 177Lu-Labeled Albumin-Binder-Conjugated PSMA-Targeting Agents with Extremely High Tumor Uptake and Enhanced Tumor-to-Kidney Absorbed Dose Ratio. J. Nucl. Med. 2020, 62, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.T.; Merkens, H.; Zhang, Z.; Uribe, C.F.; Lau, J.; Zhang, C.; Colpo, N.; Lin, K.S.; Bénard, F. Enhancing Treatment Efficacy of 177Lu-PSMA-617 with the Conjugation of an Albumin-Binding Motif: Preclinical Dosimetry and Endoradiotherapy Studies. Mol. Pharm. 2018, 15, 5183–5191. [Google Scholar] [CrossRef]
- Deberle, L.M.; Benešová, M.; Umbricht, C.A.; Borgna, F.; Büchler, M.; Zhernosekov, K.; Schibli, R.; Müller, C. Development of a new class of PSMA radioligands comprising ibuprofen as an albumin-binding entity. Theranostics 2020, 10, 1678–1693. [Google Scholar] [CrossRef]
- Baranski, A.-C.; Lindner, T.; Toennesmann, R.; Meyer, P.; Mier, W.; Eder, M. Reduction of salivary gland uptake in endoradiotherapy of prostate cancer: First preclinical data of a cleavable derivative of PSMA-617. J. Nucl. Med. 2019, 60 (Suppl. 1), 1027. [Google Scholar]
- Vaidyanathan, G.; Kang, C.M.; McDougald, D.; Minn, I.; Brummet, M.; Pomper, M.G.; Zalutsky, M.R. Brush border enzyme-cleavable linkers: Evaluation for reducing renal uptake of radiolabeled prostate-specific membrane antigen inhibitors. Nucl. Med. Biol. 2018, 62–63, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, E.A.M.; van Weerden, W.M.; Nonnekens, J.; de Jong, M. The Future of PSMA-Targeted Radionuclide Therapy: An Overview of Recent Preclinical Research. Pharmaceutics 2019, 11, 560. [Google Scholar] [CrossRef] [Green Version]
- Brandt, M.; Cardinale, J.; Giammei, C.; Guarrochena, X.; Happl, B.; Jouini, N.; Mindt, T.L. Mini-review: Targeted radiopharmaceuticals incorporating reversible, low molecular weight albumin binders. Nucl. Med. Biol. 2019, 70, 46–52. [Google Scholar] [CrossRef]
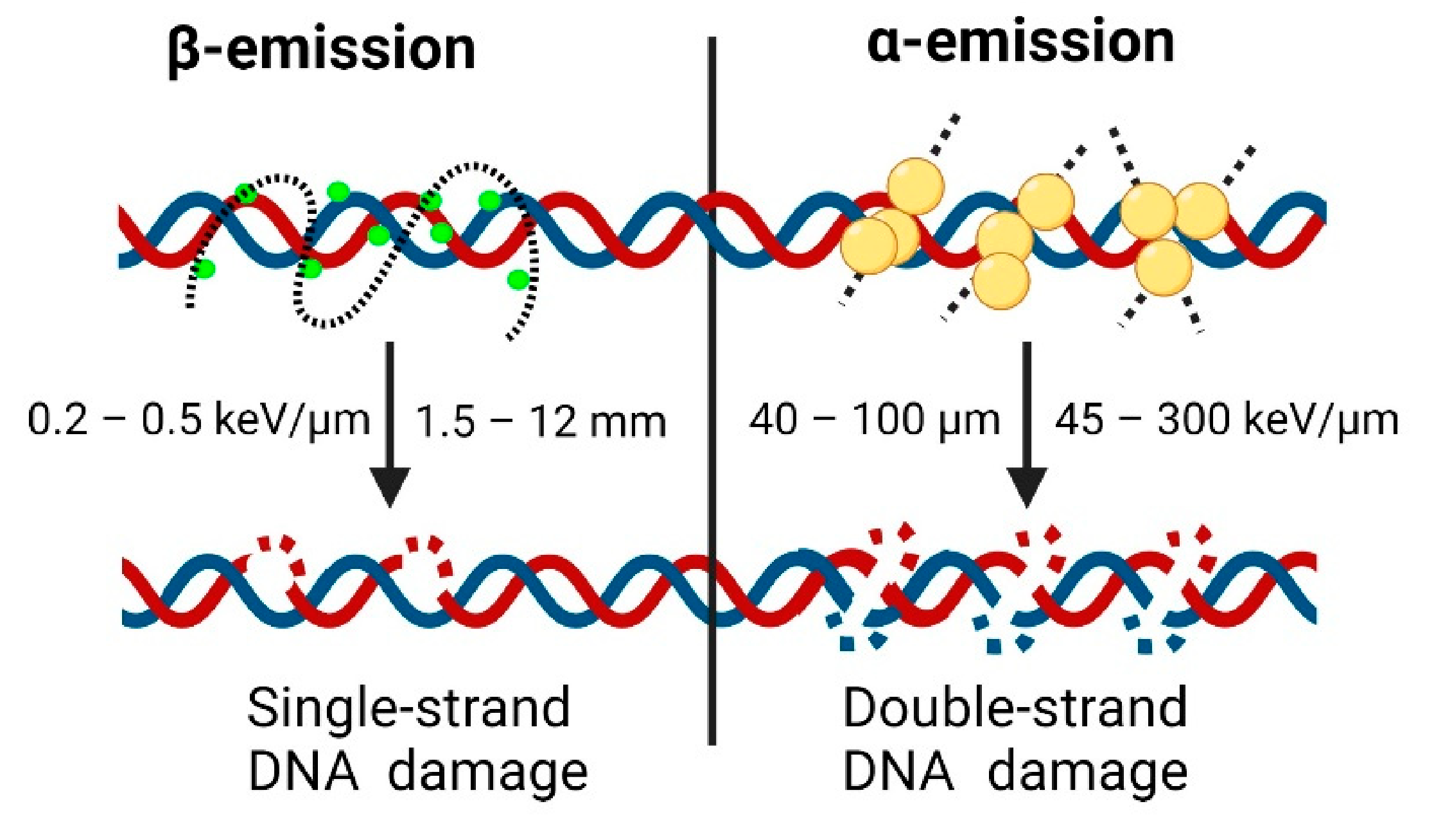
- Murshed, H. Radiation Biology. In Fundamentals of Radiation Oncology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 57–87. [Google Scholar]
- Kassis, A.I. Therapeutic Radionuclides: Biophysical and Radiobiologic Principles. Semin. Nucl. Med. 2008, 38, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Enger, S.A.; Hartman, T.; Carlsson, J.; Lundqvist, H. Cross-fire doses from β-emitting radionuclides in targeted radiotherapy. A theoretical study based on experimentally measured tumor characteristics. Phys. Med. Biol. 2008, 53, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, D.H.; Lichtenstein, M.; Better, N.; Rosenthal, M. Results of Strontium-89 Therapy in Patients With Prostate Cancer Resistant to Chemotherapy. Clin. Nucl. Med. 2004, 29, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Hao, G.; Mastren, T.; Silvers, W.; Hassan, G.; Öz, O.K.; Sun, X. Copper-67 radioimmunotheranostics for simultaneous immunotherapy and immuno-SPECT. Sci. Rep. 2021, 11, 3622. [Google Scholar] [CrossRef]
- Laboratory, B.N. Nudat 2. Available online: https://www.nndc.bnl.gov/nudat2/ (accessed on 17 May 2021).
- Riaz, A.; Awais, R.; Salem, R. Side effects of yttrium-90 radioembolization. Front. Oncol. 2014, 4, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, T.; Banerjee, S. Theranostic Applications of Lutetium-177 in Radionuclide Therapy. Curr. Radiopharm. 2015, 9, 94–101. [Google Scholar] [CrossRef]
- McCready, V.R. Radioiodine–the success story of Nuclear Medicine: 75th Anniversary of the first use of Iodine-131 in humans. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 179–182. [Google Scholar] [CrossRef] [Green Version]
- Gracheva, N.; Müller, C.; Talip, Z.; Heinitz, S.; Köster, U.; Zeevaart, J.R.; Vögele, A.; Schibli, R.; van der Meulen, N.P. Production and characterization of no-carrier-added 161Tb as an alternative to the clinically-applied 177Lu for radionuclide therapy. EJNMMI Radiopharm. Chem. 2019, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Schellhammer, P.F.; Wright, G.L. Biomolecular and clinical characteristics of PSA and other candidate prostate tumor markers. Urol. Clin. N. Am. 1993, 20, 597–606. [Google Scholar] [CrossRef]
- Smith-Jones, P.M.; Vallabhajosula, S.; St. Omer, S.; Navarro, V.; Goldsmith, S.J.; Bander, N.H. 177Lu-DOTA-HuJ591: A new radiolabeled monoclonal antibody (MAb) for targeted therapy of prostate cancer. J. Label. Compd. Radiopharm. 2001, 44, S90–S92. [Google Scholar] [CrossRef]
- Vallabhajosula, S.; Smith-Jones, P.M.; Navarro, V.; Goldsmith, S.J.; Bander, N.H. Radioimmunotherapy of Prostate Cancer in Human Xenografts Using Monoclonal Antibodies Specific to Prostate Specific Membrane Antigen (PSMA): Studies in Nude Mice. Prostate 2004, 58, 145–155. [Google Scholar] [CrossRef]
- Müller, C.; Umbricht, C.A.; Gracheva, N.; Tschan, V.J.; Pellegrini, G.; Bernhardt, P.; Zeevaart, J.R.; Köster, U.; Schibli, R.; van der Meulen, N.P. Terbium-161 for PSMA-targeted radionuclide therapy of prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1919–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zia, N.A.; Cullinane, C.; Van Zuylekom, J.K.; Waldeck, K.; McInnes, L.E.; Buncic, G.; Haskali, M.B.; Roselt, P.D.; Hicks, R.J.; Donnelly, P.S. A Bivalent Inhibitor of Prostate Specific Membrane Antigen Radiolabeled with Copper-64 with High Tumor Uptake and Retention. Angew. Chem. Int. Ed. 2019, 58, 14991–14994. [Google Scholar] [CrossRef]
- McInnes, L.E.; Cullinane, C.; Roselt, P.; Jackson, S.; Blyth, B.; van Dam, E.; Zia, N.A.; Harris, M.J.; Hicks, R.J.; Donnelly, P.S. Therapeutic Efficacy of a Bivalent Inhibitor of Prostate-Specific Membrane Antigen Labeled with Copper-67. J. Nucl. Med. 2020. [Google Scholar] [CrossRef]
- Deb, N.; Goris, M.; Trisler, K.; Fowler, S.; Saal, J.; Ning, S.; Becker, M.; Marquez, C.; Knox, S. Treatment of hormone-refractory prostate cancer with 90Y-CYT-356 monoclonal antibody. Clin. Cancer Res. 1996, 2, 1289–1297. [Google Scholar] [PubMed]
- Milowsky, M.I.; Nanus, D.M.; Kostakoglu, L.; Vallabhajosula, S.; Goldsmith, S.J.; Bander, N.H. Phase I trial of yttrium-90-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for androgen-independent prostate cancer. J. Clin. Oncol. 2004, 22, 2522–2531. [Google Scholar] [CrossRef] [PubMed]
- Vallabhajosula, S.; Goldsmith, S.J.; Kostakoglu, L.; Milowsky, M.I.; Nanus, D.M.; Bander, N.H. Radioimmunotherapy of prostate cancer using 90Y- and 177Lu-labeled J591 monoclonal antibodies: Effect of multiple treatments on myelotoxicity. Clin. Cancer Res. 2005, 11, 7195s–7200s. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, S.T.; Milowsky, M.I.; Morris, M.J.; Vallabhajosula, S.; Goldsmith, S.; Matulich, D.; Kaplan, J.; Berger, F.; Scher, H.I.; Bander, N.H.; et al. Phase II trial of 177Lutetium radiolabeled anti-prostate-specific membrane antigen (PSMA) monoclonal antibody J591 (177Lu- J591) in patients (pts) with metastatic castrate-resistant prostate cancer (metCRPC). J. Clin. Oncol. 2008, 26, 5140. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Vallabhajosula, S.; Osborne, J.; Goldsmith, S.J.; Petrillo, K.; Tyrell, L.; Dhillon, G.S.; Beltran, H.; Bander, N.H.; Nanus, D.M. Phase I trial of fractionated-dose 177lutetium radiolabeled anti-prostate-specific membrane antigen (PSMA) monoclonal antibody J591 (177Lu-J591) in patients (pts) with metastatic castration-resistant prostate cancer (metCRPC). J. Clin. Oncol. 2010, 28, 4667. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Whang, Y.E.; Kaur, G.; Vallabhajosula, S.; Christos, P.J.; Nikolopoulou, A.; Jhanwar, Y.; Sheikh, A.; Ireland, A.; Garcias-Espana, C.; et al. Phase I trial of docetaxel/prednisone plus fractionated dose radiolabeled anti-prostate-specific membrane antigen (PSMA) monoclonal antibody 177Lu-J591 in patients with metastatic, castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2014, 32, 5064. [Google Scholar] [CrossRef]
- Afshar-Oromieh, A.; Haberkorn, U.; Zechmann, C.; Armor, T.; Mier, W.; Spohn, F.; Debus, N.; Holland-Letz, T.; Babich, J.; Kratochwil, C. Repeated PSMA-targeting radioligand therapy of metastatic prostate cancer with 131I-MIP-1095. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 950–959. [Google Scholar] [CrossRef] [Green Version]
- Kratochwil, C.; Giesel, F.L.; Eder, M.; Afshar-Oromieh, A.; Benešová, M.; Mier, W.; Kopka, K.; Haberkorn, U. [177Lu]Lutetium-labelled PSMA ligand-induced remission in a patient with metastatic prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 987–988. [Google Scholar] [CrossRef]
- Ahmadzadehfar, H.; Rahbar, K.; Kürpig, S.; Bögemann, M.; Claesener, M.; Eppard, E.; Gärtner, F.; Rogenhofer, S.; Schäfers, M.; Essler, M. Early side effects and first results of radioligand therapy with 177Lu-DKFZ-617 PSMA of castrate-resistant metastatic prostate cancer: A two-centre study. EJNMMI Res. 2015, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzadehfar, H.; Eppard, E.; Kürpig, S.; Fimmers, R.; Yordanova, A.; Schlenkhoff, C.D.; Gärtner, F.; Rogenhofer, S.; Essler, M. Therapeutic response and side effects of repeated radioligand therapy with 177Lu-PSMA-DKFZ-617 of castrate-resistant metastatic prostate cancer. Oncotarget 2016, 7, 12477–12488. [Google Scholar] [CrossRef]
- Rahbar, K.; Schmidt, M.; Heinzel, A.; Eppard, E.; Bode, A.; Yordanova, A.; Claesener, M.; Ahmadzadehfar, H. Response and tolerability of a single dose of 177Lu-PSMA-617 in patients with metastatic castration-resistant prostate cancer: A multicenter retrospective analysis. J. Nucl. Med. 2016, 57, 1334–1338. [Google Scholar] [CrossRef] [Green Version]
- Rahbar, K.; Ahmadzadehfar, H.; Kratochwil, C.; Haberkorn, U.; Schafers, M.; Essler, M.; Baum, R.P.; Kulkarni, H.R.; Schmidt, M.; Drzezga, A.; et al. German multicenter study investigating 177Lu-PSMA-617 Radioligand therapy in advanced prostate cancer patients. J. Nucl. Med. 2017, 58, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzadehfar, H.; Wegen, S.; Yordanova, A.; Fimmers, R.; Kürpig, S.; Eppard, E.; Wei, X.; Schlenkhoff, C.; Hauser, S.; Essler, M. Overall survival and response pattern of castration-resistant metastatic prostate cancer to multiple cycles of radioligand therapy using [177Lu]Lu-PSMA-617. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1448–1454. [Google Scholar] [CrossRef]
- Rahbar, K.; Boegemann, M.; Yordanova, A.; Eveslage, M.; Schäfers, M.; Essler, M.; Ahmadzadehfar, H. PSMA targeted radioligandtherapy in metastatic castration resistant prostate cancer after chemotherapy, abiraterone and/or enzalutamide. A retrospective analysis of overall survival. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 12–19. [Google Scholar] [CrossRef]
- Hofman, M.S.; Violet, J.; Hicks, R.J.; Ferdinandus, J.; Ping Thang, S.; Akhurst, T.; Iravani, A.; Kong, G.; Ravi Kumar, A.; Murphy, D.G.; et al. [177Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): A single-centre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 825–833. [Google Scholar] [CrossRef]
- Yordanova, A.; Linden, P.; Hauser, S.; Meisenheimer, M.; Kürpig, S.; Feldmann, G.; Gaertner, F.C.; Essler, M.; Ahmadzadehfar, H. Outcome and safety of rechallenge [177Lu]Lu-PSMA-617 in patients with metastatic prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1073–1080. [Google Scholar] [CrossRef]
- Maffey-Steffan, J.; Scarpa, L.; Svirydenka, A.; Nilica, B.; Mair, C.; Buxbaum, S.; Bektic, J.; von Guggenberg, E.; Uprimny, C.; Horninger, W.; et al. The 68Ga/177Lu-theragnostic concept in PSMA-targeting of metastatic castration–resistant prostate cancer: Impact of post-therapeutic whole-body scintigraphy in the follow-up. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 695–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasul, S.; Hacker, M.; Kretschmer-Chott, E.; Leisser, A.; Grubmüller, B.; Kramer, G.; Shariat, S.; Wadsak, W.; Mitterhauser, M.; Hartenbach, M.; et al. Clinical outcome of standardized 177Lu-PSMA-617 therapy in metastatic prostate cancer patients receiving 7400 MBq every 4 weeks. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Rupp, N.J.; Umbricht, C.A.; Pizzuto, D.A.; Lenggenhager, D.; Töpfer, A.; Müller, J.; Muehlematter, U.J.; Ferraro, D.A.; Messerli, M.; Morand, G.B.; et al. First clinicopathologic evidence of a non-PSMA-related uptake mechanism for 68Ga-PSMA-11 in salivary glands. J. Nucl. Med. 2019, 60, 1270–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein Nulent, T.J.W.; Valstar, M.H.; de Keizer, B.; Willems, S.M.; Smit, L.A.; Al-Mamgani, A.; Smeele, L.E.; van Es, R.J.J.; de Bree, R.; Vogel, W.V. Physiologic distribution of PSMA-ligand in salivary glands and seromucous glands of the head and neck on PET/CT. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2018, 125, 478–486. [Google Scholar] [CrossRef]
- Tönnesmann, R.; Meyer, P.T.; Eder, M.; Baranski, A.C. [177Lu]Lu-PSMA-617 salivary gland uptake characterized by quantitative in vitro autoradiography. Pharmaceuticals 2019, 12, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov Lu177-EB-PSMA617 Radionuclide Treatment in Patients with Metastatic Castration-Resistant Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03780075?term=NCT03780075&draw=2&rank=1 (accessed on 17 May 2021).
- ClinicalTrials.gov 177Lu Radiolabeled Monoclonal Antibody HuJ591 (177Lu-J591) and Ketoconazole in Patients with Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00859781?term=NCT00859781&draw=2&rank=1 (accessed on 17 May 2021).
- ClinicalTrials.gov A Trial of 177Lu-PSMA617 Theranostic Versus Cabazitaxel in Progressive Metastatic Castration Resistant Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03392428?term=NCT03392428&draw=2&rank=1 (accessed on 17 May 2021).
- ClinicalTrials.gov Study Evaluating mCRPC Treatment Using PSMA [Lu-177]-PNT2002 Therapy after Second-line Hormonal Treatment. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04647526 (accessed on 9 June 2021).
- Graf, F.; Fahrer, J.; Maus, S.; Morgenstern, A.; Bruchertseifer, F.; Venkatachalam, S.; Fottner, C.; Weber, M.M.; Huelsenbeck, J.; Schreckenberger, M.; et al. DNA double strand breaks as predictor of efficacy of the alpha-particle emitter Ac-225 and the electron emitter Lu-177 for somatostatin receptor targeted radiotherapy. PLoS ONE 2014, 9, e88239. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, R.; Siamof, C.M.; Dash, A.; Cai, W. Targeted α-therapy of prostate cancer using radiolabeled PSMA inhibitors: A game changer in nuclear medicine. Am. J. Nucl. Med. Mol. Imaging 2018, 8, 247–267. [Google Scholar] [PubMed]
- van der Doelen, M.J.; Mehra, N.; van Oort, I.M.; Looijen-Salamon, M.G.; Janssen, M.J.R.; Custers, J.A.E.; Slootbeek, P.H.J.; Kroeze, L.I.; Bruchertseifer, F.; Morgenstern, A.; et al. Clinical outcomes and molecular profiling of advanced metastatic castration-resistant prostate cancer patients treated with 225Ac-PSMA-617 targeted alpha-radiation therapy. Urol. Oncol. Semin. Orig. Investig. 2020. [Google Scholar] [CrossRef]
- De Vincentis, G.; Gerritsen, W.; Gschwend, J.E.; Hacker, M.; Lewington, V.; O’Sullivan, J.M.; Oya, M.; Pacilio, M.; Parker, C.; Shore, N.; et al. Advances in Targeted Alpha Therapy for Prostate Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1728–1739. [Google Scholar] [CrossRef] [PubMed]
- Wilbur, D. [211At]Astatine-Labeled Compound Stability: Issues with Released [211At]Astatide and Development of Labeling Reagents to Increase Stability. Curr. Radiopharm. 2010, 1, 144–176. [Google Scholar] [CrossRef]
- Klusa, D.; Lohaus, F.; Furesi, G.; Rauner, M.; Benešová, M.; Krause, M.; Kurth, I.; Peitzsch, C. Metastatic Spread in Prostate Cancer Patients Influencing Radiotherapy Response. Front. Oncol. 2021, 10, 1–27. [Google Scholar] [CrossRef]
- Juzeniene, A.; Stenberg, V.Y.; Bruland, Ø.S.; Larsen, R.H. Preclinical and Clinical Status of PSMA-Targeted Alpha Therapy for Metastatic Castration-Resistant Prostate Cancer. Cancers 2021, 13, 779. [Google Scholar] [CrossRef]
- Deshayes, E.; Roumiguie, M.; Thibault, C.; Beuzeboc, P.; Cachin, F.; Hennequin, C.; Huglo, D.; Rozet, F.; Kassab-Chahmi, D.; Rebillard, X.; et al. Radium 223 dichloride for prostate cancer treatment. Drug Des. Devel. Ther. 2017, 11, 2643–2651. [Google Scholar] [CrossRef] [Green Version]
- Ahenkorah, S.; Cassells, I.; Deroose, C.M.; Cardinaels, T.; Burgoyne, A.R.; Bormans, G.; Ooms, M.; Cleeren, F. Bismuth-213 for targeted radionuclide therapy: From atom to bedside. Pharmaceutics 2021, 13, 599. [Google Scholar] [CrossRef] [PubMed]
- de Kruijff, R.M.; Wolterbeek, H.T.; Denkova, A.G. A critical review of alpha radionuclide therapy-how to deal with recoiling daughters? Pharmaceuticals 2015, 8, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Lindegren, S.; Albertsson, P.; Bäck, T.; Jensen, H.; Palm, S.; Aneheim, E. Realizing Clinical Trials with Astatine-211: The Chemistry Infrastructure. Cancer Biother. Radiopharm. 2020, 35, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Ayed, T.; Pilmé, J.; Tézé, D.; Bassal, F.; Barbet, J.; Chérel, M.; Champion, J.; Maurice, R.; Montavon, G.; Galland, N. 211At-labeled agents for alpha-immunotherapy: On the in vivo stability of astatine-agent bonds. Eur. J. Med. Chem. 2016, 116, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Zalutsky, M.; Vaidyanathan, G. Astatine-211-Labeled Radiotherapeutics An Emerging Approach to Targeted Alpha-Particle Radiotherapy. Curr. Pharm. Des. 2005, 6, 1433–1455. [Google Scholar] [CrossRef]
- Turkington, T.G.; Zalutsky, M.R.; Jaszczak, R.J.; Garg, P.K.; Vaidyanathan, G.; Coleman, R.E. Measuring astatine-211 distributions with SPECT. Phys. Med. Biol. 1993, 38, 1121–1130. [Google Scholar] [CrossRef]
- Yong, K.; Brechbiel, M.W. Towards translation of 212Pb as a clinical therapeutic; Getting the lead in! Dalt. Trans. 2011, 40, 6068–6076. [Google Scholar] [CrossRef]
- Müller, C.; Vermeulen, C.; Köster, U.; Johnston, K.; Türler, A.; Schibli, R.; van der Meulen, N.P. Alpha-PET with terbium-149: Evidence and perspectives for radiotheragnostics. EJNMMI Radiopharm. Chem. 2017, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- George, D.J.; McDevitt, M.R.; Barendswaard, E.; Ma, D.; Lai, L.; Curcio, M.J.; Sgouros, G.; Ballangrud, A.M.; Yang, W.H.; Finn, R.D.; et al. An α-particle emitting antibody ([213Bi]J591) for radioimmmunotherapy of prostate cancer. Prostate J. 2001, 3, 1. [Google Scholar] [CrossRef]
- Li, Y.; Tian, Z.; Rizvi, S.M.A.; Bander, N.H.; Allen, B.J. In vitro and preclinical targeted alpha therapy of human prostate cancer with Bi-213 labeled J591 antibody against the prostate specific membrane antigen. Prostate Cancer Prostatic Dis. 2002, 5, 36–46. [Google Scholar] [CrossRef]
- Preston Campbell, J.; Merkel, A.R.; Kathryn Masood-Campbell, S.; Elefteriou, F.; Sterling, J.A. Models of bone metastasis. J. Vis. Exp. 2012. [Google Scholar] [CrossRef]
- Wilbur, D.S.; Hamlin, D.; Nguyen, H.; Nakamae, H.; Chyan, M.-K.; Vessella, R.; Sandmaier, B. Preliminary studies using At-211-labeled anti-PSMA mAb for treatment of metastatic prostate cancer in a mouse model. J. Nucl. Med. 2009, 50, 39. [Google Scholar]
- Wilbur, D.S.; Chyan, M.K.; Hamlin, D.K.; Nguyen, H.; Vessella, R.L. Reagents for astatination of biomolecules. 5. Evaluation of hydrazone linkers in 211At-and 125I-labeled closo-Decaborate(2-) conjugates of Fab as a means of decreasing kidney retention. Bioconjug. Chem. 2011, 22, 1089–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mease, R.C.; Kang, C.; Kumar, V.; Ray, S.; Minn, I.; Brummet, M.; Gabrielson, K.; Feng, Y.; Park, A.; Kiess, A.; et al. An improved 211At-labeled agent for PSMA-targeted alpha therapy. J. Nucl. Med. 2021. ahead of print. [Google Scholar] [CrossRef]
- Umbricht, C.A.; Köster, U.; Bernhardt, P.; Gracheva, N.; Johnston, K.; Schibli, R.; van der Meulen, N.P.; Müller, C. Alpha-PET for Prostate Cancer: Preclinical investigation using 149Tb-PSMA-617. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- dos Santos, J.C.; Schäfer, M.; Bauder-Wüst, U.; Lehnert, W.; Leotta, K.; Morgenstern, A.; Kopka, K.; Haberkorn, U.; Mier, W.; Kratochwil, C. Development and dosimetry of 203Pb/212Pb-labelled PSMA ligands: Bringing “the lead” into PSMA-targeted alpha therapy? Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Stenberg, V.Y.; Juzeniene, A.; Chen, Q.; Yang, X.; Bruland, Ø.S.; Larsen, R.H. Preparation of the alpha-emitting prostate-specific membrane antigen targeted radioligand [212Pb]Pb-NG001 for prostate cancer. J. Label. Compd. Radiopharm. 2020, 63, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.R.; Minn, I.; Kumar, V.; Josefsson, A.; Lisok, A.; Brummet, M.; Chen, J.; Kiess, A.P.; Baidoo, K.; Brayton, C.; et al. Preclinical evaluation of 203/212Pb-labeled low-molecular-weight compounds for targeted radiopharmaceutical therapy of prostate cancer. J. Nucl. Med. 2020, 61, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Novartis Announces Positive Result of Phase III Study with Radioligand Therapy 177Lu-PSMA-617 in Patients with Advanced Prostate Cancer. Available online: https://www.novartis.com/news/media-releases/novartis-announces-positive-result-phase-iii-study-radioligand-therapy-177lu-psma-617-patients-advanced-prostate-cancer (accessed on 30 May 2021).
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.L.; Weis, M.; Verburg, F.A.; Mottaghy, F.; Kopka, K.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. 225Ac-PSMA-617 for PSMA-targeted α-radiation therapy of metastatic castration-resistant prostate cancer. J. Nucl. Med. 2016, 57, 1941–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Hohenfellner, M.; Giesel, F.L.; Haberkorn, U.; Morgenstern, A. Targeted a-therapy of metastatic castration-resistant prostate cancer with 225Ac-PSMA-617: Swimmer-Plot Analysis Suggests efficacy regarding duration of tumor control. J. Nucl. Med. 2018, 59, 795–802. [Google Scholar] [CrossRef] [Green Version]
- de Medeiros, R.B.; Grigolon, M.V.; Araújo, T.P.; Srougi, M. Câncer de próstata metastático resistente à castração (mCRPC) tratado com 225Ac-PSMA-617. Relato de caso. Brazilian J. Oncol. 2019. [Google Scholar] [CrossRef]
- Yadav, M.P.; Ballal, S.; Sahoo, R.K.; Tripathi, M.; Seth, A.; Bal, C. Efficacy and safety of 225Ac-PSMA-617 targeted alpha therapy in metastatic castration-resistant prostate cancer patients. Theranostics 2020, 10, 9364–9377. [Google Scholar] [CrossRef]
- Feuerecker, B.; Tauber, R.; Knorr, K.; Heck, M.; Beheshti, A.; Seidl, C.; Bruchertseifer, F.; Pickhard, A.; Gafita, A.; Kratochwil, C.; et al. Activity and Adverse Events of Actinium-225-PSMA-617 in Advanced Metastatic Castration-resistant Prostate Cancer After Failure of Lutetium-177-PSMA [Formula presented]. Eur. Urol. 2021, 79, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Sathekge, M.; Bruchertseifer, F.; Knoesen, O.; Reyneke, F.; Lawal, I.; Lengana, T.; Davis, C.; Mahapane, J.; Corbett, C.; Vorster, M.; et al. 225Ac-PSMA-617 in chemotherapy-naive patients with advanced prostate cancer: A pilot study. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 129–138. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov Phase I Trial of 225Ac-J591 in Patients with mCRPC. Available online: https://clinicaltrials.gov/ct2/show/NCT03276572?term=225Ac-J591&draw=2&rank=2 (accessed on 4 May 2021).
- ClinicalTrials.gov Fractionated and Multiple Dose 225Ac-J591 for Progressive mCRPC. Available online: https://clinicaltrials.gov/ct2/show/NCT04506567?term=225Ac-J591&draw=2&rank=3 (accessed on 4 May 2021).
- ClinicalTrials.gov Re-treatment 225Ac-J591 for mCRPC. Available online: https://clinicaltrials.gov/ct2/show/NCT04576871?term=225Ac-J591&draw=2&rank=1 (accessed on 4 May 2021).
- Hammer, S.; Larssen, A.; Ellingsen, C.; Geraudie, S.; Grant, D.; Indrevoll, B.; von Ahsen, O.; Kristian, A.; Hagemann, U.B.; Karlsson, J.; et al. Preclinical pharmacology of the PSMA-targeted thorium-227 conjugate PSMA-TTC: A novel targeted alpha therapeutic for the treatment of prostate cancer. Clin. Cancer Res. 2017, 77, 5200. [Google Scholar] [CrossRef]
- Hammer, S.; Hagemann, U.B.; Zitzmann-Kolbe, S.; Larsen, A.; Ellingsen, C.; Geraudie, S.; Grant, D.; Indrevoll, B.; Smeets, R.; Von Ahsen, O.; et al. Preclinical efficacy of a PSMA-targeted thorium-227 conjugate (PSMA-TTC), a targeted alpha therapy for prostate cancer. Clin. Cancer Res. 2020, 26, 1985–1996. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Anti-Tumor Activity of a Thorium-227 Labeled Antibody-Chelator Conjugate, in Patients with Metastatic Castration Resistant Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03724747 (accessed on 4 May 2021).
- Smits, M.; Gerritsen, W.; Mehra, N. Future therapeutic strategies for metastatic prostate cancer. Tijdschr. Voor Urol. 2019, 9, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Palacios, D.A.; Miyake, M.; Rosser, C.J. Radiosensitization in prostate cancer: Mechanisms and targets. BMC Urol. 2013, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesson, M.; Rae, C.; Nixon, C.; Babich, J.W.; Mairs, R.J. Preliminary evaluation of prostate-targeted radiotherapy using 131I-MIP-1095 in combination with radiosensitising chemotherapeutic drugs. J. Pharm. Pharmacol. 2016, 68, 912–921. [Google Scholar] [CrossRef]
- Crumbaker, M.; Pathmanandavel, S.; Yam, A.O.; Nguyen, A.; Ho, B.; Chan, L.; Ende, J.A.; Rofe, C.; Kongrak, K.; Kwan, E.M.; et al. Phase I/II Trial of the Combination of 177Lutetium Prostate specific Membrane Antigen 617 and Idronoxil (NOX66) in Men with End-stage Metastatic Castration-resistant Prostate Cancer (LuPIN). Eur. Urol. Oncol. 2020. [Google Scholar] [CrossRef]
- Emmett, L.; Pathmanandavel, S.; Crumbaker, M.; Rofe, C.; Yam, A.O.W.; Ho, B.; Chan, W.L.; Sharma, S.; Keane, J.; Hickey, A.J.; et al. Updated results of a phase I/II prospective dose escalation trial evaluating safety and efficacy of combination 177Lu PSMA 617 and idronoxil in men with mCRPC post androgen signalling inhibition and taxane chemotherapy (LuPIN trial). J. Clin. Oncol. 2020, 38, 5557. [Google Scholar] [CrossRef]
- Kumar, P. A new paradigm for the treatment of high-risk prostate cancer: Radiosensitization with docetaxel. Rev. Urol. 2003, 5 (Suppl. 3), S71–S77. [Google Scholar]
- Kelly, M.P.; Lee, S.T.; Lee, F.T.; Smyth, F.E.; Davis, I.D.; Brechbiel, M.W.; Scott, A.M. Therapeutic efficacy of 177Lu-CHX-A″-DTPA-hu3SI93 radioimmunotherapy in prostate cancer is enhanced by EGFR inhibition or docetaxel chemotherapy. Prostate 2009, 69, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Batra, J.S.; Niaz, M.J.; Whang, Y.E.; Sheikh, A.; Thomas, C.; Christos, P.; Vallabhajosula, S.; Jhanwar, Y.S.; Molina, A.M.; Nanus, D.M.; et al. Phase I trial of docetaxel plus lutetium-177-labeled anti–prostate-specific membrane antigen monoclonal antibody J591 (177Lu-J591) for metastatic castration-resistant prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 848.e9–848.e16. [Google Scholar] [CrossRef]
- Dhiantravan, N.; Emmett, L.; Joshua, A.M.; Pattison, D.A.; Francis, R.J.; Williams, S.; Sandhu, S.; Davis, I.D.; Vela, I.; Neha, N.; et al. UpFrontPSMA: A randomized phase 2 study of sequential 177Lu-PSMA-617 and docetaxel vs docetaxel in metastatic hormone-naïve prostate cancer (clinical trial protocol). BJU Int. 2021. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, M.; Heslop, L.; Govender, T.; Korowlay, N.; Singh, A.; Choudhary, P.; Sathekge, M. The Outcome and Safety of Re-challenge Lutetium-177 PSMA (177Lu-PSMA) Therapy with Low-Dose Docetaxel as a Radiosensitizer—a Promising Combination in Metastatic Castrate-Resistant Prostate Cancer (mCRPC): A Case Report. Nucl. Med. Mol. Imaging 2021, 55, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Langbein, T.; Chaussé, G.; Baum, R.P. Salivary gland toxicity of PSMA radioligand therapy: Relevance and preventive strategies. J. Nucl. Med. 2018, 59, 1172–1173. [Google Scholar] [CrossRef] [Green Version]
- Kratochwil, C.; Giesel, F.L.; Stefanova, M.; Benesova, M.; Bronzel, M.; Afshar-Oromieh, A.; Mier, W.; Eder, M.; Kopka, K.; Haberkorn, U. PSMA-targeted radionuclide therapy of metastatic castration-resistant prostate cancer with 177Lu-Labeled PSMA-617. J. Nucl. Med. 2016, 57, 1170–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, B.; Nisli, S.; Ergul, N.; Gursu, R.U.; Acikgoz, O.; Cxermik, T.F. Effect of external cooling on 177Lu-PSMA uptake by the parotid glands. J. Nucl. Med. 2019, 60, 1388–1393. [Google Scholar] [CrossRef] [Green Version]
- Baum, R.P.; Langbein, T.; Singh, A.; Shahinfar, M.; Schuchardt, C.; Volk, G.F.; Kulkarni, H. Injection of Botulinum Toxin for Preventing Salivary Gland Toxicity after PSMA Radioligand Therapy: An Empirical Proof of a Promising Concept. Nucl. Med. Mol. Imaging 2018, 52, 80–81. [Google Scholar] [CrossRef]
- Rathke, H.; Kratochwil, C.; Hohenberger, R.; Giesel, F.L.; Bruchertseifer, F.; Flechsig, P.; Morgenstern, A.; Hein, M.; Plinkert, P.; Haberkorn, U.; et al. Initial clinical experience performing sialendoscopy for salivary gland protection in patients undergoing 225Ac-PSMA-617 RLT. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 139–147. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Leotta, K.; Eder, M.; Hoppe-Tich, T.; Youssoufian, H.; Kopka, K.; Babich, J.W.; Haberkorn, U. PMPA for nephroprotection in PSMA-targeted radionuclide therapy of prostate cancer. J. Nucl. Med. 2015, 56, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vornov, J.J.; Peters, D.; Nedelcovych, M.; Hollinger, K.; Rais, R.; Slusher, B.S. Looking for Drugs in All the Wrong Places: Use of GCPII Inhibitors Outside the Brain. Neurochem. Res. 2020, 45, 1256–1267. [Google Scholar] [CrossRef]
- Kalidindi, T.M.; Lee, S.G.; Jou, K.; Chakraborty, G.; Skafida, M.; Tagawa, S.T.; Bander, N.H.; Schoder, H.; Bodei, L.; Pandit-Taskar, N.; et al. A simple strategy to reduce the salivary gland and kidney uptake of PSMA-targeting small molecule radiopharmaceuticals. Eur. J. Nucl. Med. Mol. Imaging 2021. [Google Scholar] [CrossRef] [PubMed]
- Harsini, S.; Saprunoff, H.; Alden, T.; Mohammadi, B.; Wilson, D.; Bénard, F. The Effects of Monosodium Glutamate on PSMA Radiotracer Uptake in Men with Recurrent Prostate Cancer: A Prospective, Randomized, Double-Blind, Placebo-Controlled Intraindividual Imaging Study. J. Nucl. Med. 2021, 62, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Paganelli, G.; Sarnelli, A.; Severi, S.; Sansovini, M.; Belli, M.L.; Monti, M.; Foca, F.; Celli, M.; Nicolini, S.; Tardelli, E.; et al. Dosimetry and safety of 177Lu PSMA-617 along with polyglutamate parotid gland protector: Preliminary results in metastatic castration-resistant prostate cancer patients. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 3008–3017. [Google Scholar] [CrossRef]
- Sarnelli, A.; Belli, M.L.; Di Iorio, V.; Mezzenga, E.; Celli, M.; Severi, S.; Tardelli, E.; Nicolini, S.; Oboldi, D.; Uccelli, L.; et al. Dosimetry of 177Lu-PSMA-617 after mannitol infusion and glutamate tablet administration: Preliminary results of EUDRACT/RSO 2016-002732-32 IRST protocol. Molecules 2019, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grønhøj, C.; Jensen, D.H.; Vester-Glowinski, P.; Jensen, S.B.; Bardow, A.; Oliveri, R.S.; Fog, L.M.; Specht, L.; Thomsen, C.; Darkner, S.; et al. Safety and Efficacy of Mesenchymal Stem Cells for Radiation-Induced Xerostomia: A Randomized, Placebo-Controlled Phase 1/2 Trial (MESRIX). Int. J. Radiat. Oncol. Biol. Phys. 2018, 101, 581–592. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radionuclide | Half-Life | Emission | Eβ(max)/Range (Max) |
|---|---|---|---|
| 67Cu | 61.9 h | β-/γ | 575 keV/2.1 mm |
| 89Sr | 50.5 d | β- | 1491 keV/7.0 mm |
| 90Y | 64.1 h | β- | 2284 keV/11.3 mm |
| 131I | 8.0 d | β-/γ | 606 keV/2.1 mm |
| 161Tb | 6.9 d | β-/Auger/CE | 150 keV/0.1 mm |
| 177Lu | 6.7 d | β-/γ | 497 keV/1.8 mm |
| Year | Num. of Patients | % with PSA Decline > 50% | Mean Administered Dose per Cycle/s | Authors and References |
|---|---|---|---|---|
| 2015 | 10 | 50% | 5.6 GBq (1 cycle) | Ahmadzadehfar et al. [135] |
| 2016 | 24 | 42% | 6.0 GBq (2 cycles) | Ahmadzadehfar et al. [136] |
| 2016 | 74 | 31% | 5.9 GBq (1 cycle) | Rahbar et al. [137] |
| 2017 | 99 | 40% | 5.9 GBq (1–4 cycles) | Rahbar et al. [138] |
| 2017 | 52 | 44% | 6.0 GBq (3–6 cycles) | Ahmadzadehfar et al. [139] |
| 2018 | 104 | 33% | 6.1 GBq (1–8 cycles) | Rahbar et al. [140] |
| 2018 | 30 | 57% | 7.5 GBq (2–4 cycles) | Hofman et al. [141] |
| 2019 | 30 | 23% | 8 GBq (1–6 cycles) | Yordanova et al. [142] |
| 2019 | 32 | 38% | 6 GBq (2–6 cycles) | Maffey-Steffan et al. [143] |
| 2020 | 54 | 58% | 7.5 GBq (3 cycles) | Rasul et al. [144] |
| 2021 | 385 | 46% | 7.4 GBq (4–6 cycles) | Sartor et al. [145] |
| Radionuclide | Half-life | Emission | Eα(max)/Range(Max) |
|---|---|---|---|
| 149Tb | 4.1 h | α/β+ | 3.97 MeV/28 µm |
| 211At | 7.2 h | α | 6.79 MeV/60 µm |
| 212Pb | 10.6 h | β- to α 212Bi | 6.05 MeV/80 µm |
| 213Bi | 45.6 min | α/β- | 8.32 MeV/84 µm |
| 223Ra | 11.4 d | α | 5.64 MeV/45 µm |
| 225Ac | 10.0 d | α/β- | 6.83 MeV/61 µm |
| 227Th | 18.7 d | α | 6.14 MeV/100 µm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Fakiri, M.; Geis, N.M.; Ayada, N.; Eder, M.; Eder, A.-C. PSMA-Targeting Radiopharmaceuticals for Prostate Cancer Therapy: Recent Developments and Future Perspectives. Cancers 2021, 13, 3967. https://doi.org/10.3390/cancers13163967
El Fakiri M, Geis NM, Ayada N, Eder M, Eder A-C. PSMA-Targeting Radiopharmaceuticals for Prostate Cancer Therapy: Recent Developments and Future Perspectives. Cancers. 2021; 13(16):3967. https://doi.org/10.3390/cancers13163967
Chicago/Turabian StyleEl Fakiri, Mohamed, Nicolas M. Geis, Nawal Ayada, Matthias Eder, and Ann-Christin Eder. 2021. "PSMA-Targeting Radiopharmaceuticals for Prostate Cancer Therapy: Recent Developments and Future Perspectives" Cancers 13, no. 16: 3967. https://doi.org/10.3390/cancers13163967
APA StyleEl Fakiri, M., Geis, N. M., Ayada, N., Eder, M., & Eder, A. -C. (2021). PSMA-Targeting Radiopharmaceuticals for Prostate Cancer Therapy: Recent Developments and Future Perspectives. Cancers, 13(16), 3967. https://doi.org/10.3390/cancers13163967