
Hyaluronic Acid-Functionalized Nanomicelles Enhance SAHA Efficacy in 3D Endometrial Cancer Models

 ,
,  ,
,  ,
,  and
and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. 2D Cell Culture
2.2. 3D Cell Culture
2.3. Confocal Laser Microscopy Analysis
2.4. Synthesis of Thiolated HA (HA-SH)
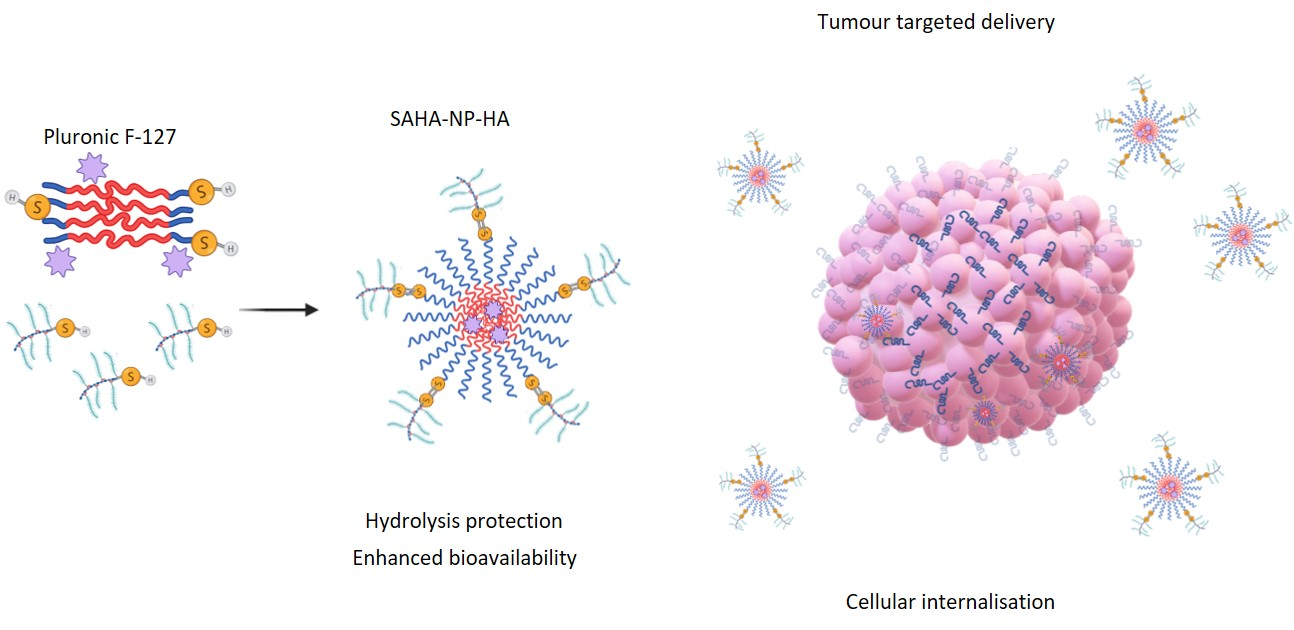
2.5. Synthesis of Pluronic® F-127 Micelles
2.6. Activation of Pluronic F127
2.7. Synthesis of Pyridyl Disulphide Ligand (2-(2-Pyridyldithio) Ethylamine)
2.8. Synthesis of F127 Pyridyl Disulphide Derivative (NP-S-S-Py)
2.9. Size and Zeta Potential Analysis with Dynamic Light Scattering (DLS)
2.10. Atomic Force Microscope (AFM)
2.11. HPLC-UV Quantification
2.12. Drug Release Profile
2.13. Cellular Penetration
2.14. Chromatin Immunoprecipitation (ChIP)
2.15. 2D/3D Cell Viability Assays
2.16. Protein Blotting and Antibodies
2.17. Immunofluorescence and Antibodies—2D
2.18. Immunofluorescence—3D
2.19. Statistical Analysis
3. Results and Discussion
3.1. Effective SAHA Dose Determination for Type I and II Endometrial Cancer Models
3.2. Physicochemical Characterisation of Functionalized and SAHA-Loaded Pluronic® F-127 Nanoparticles
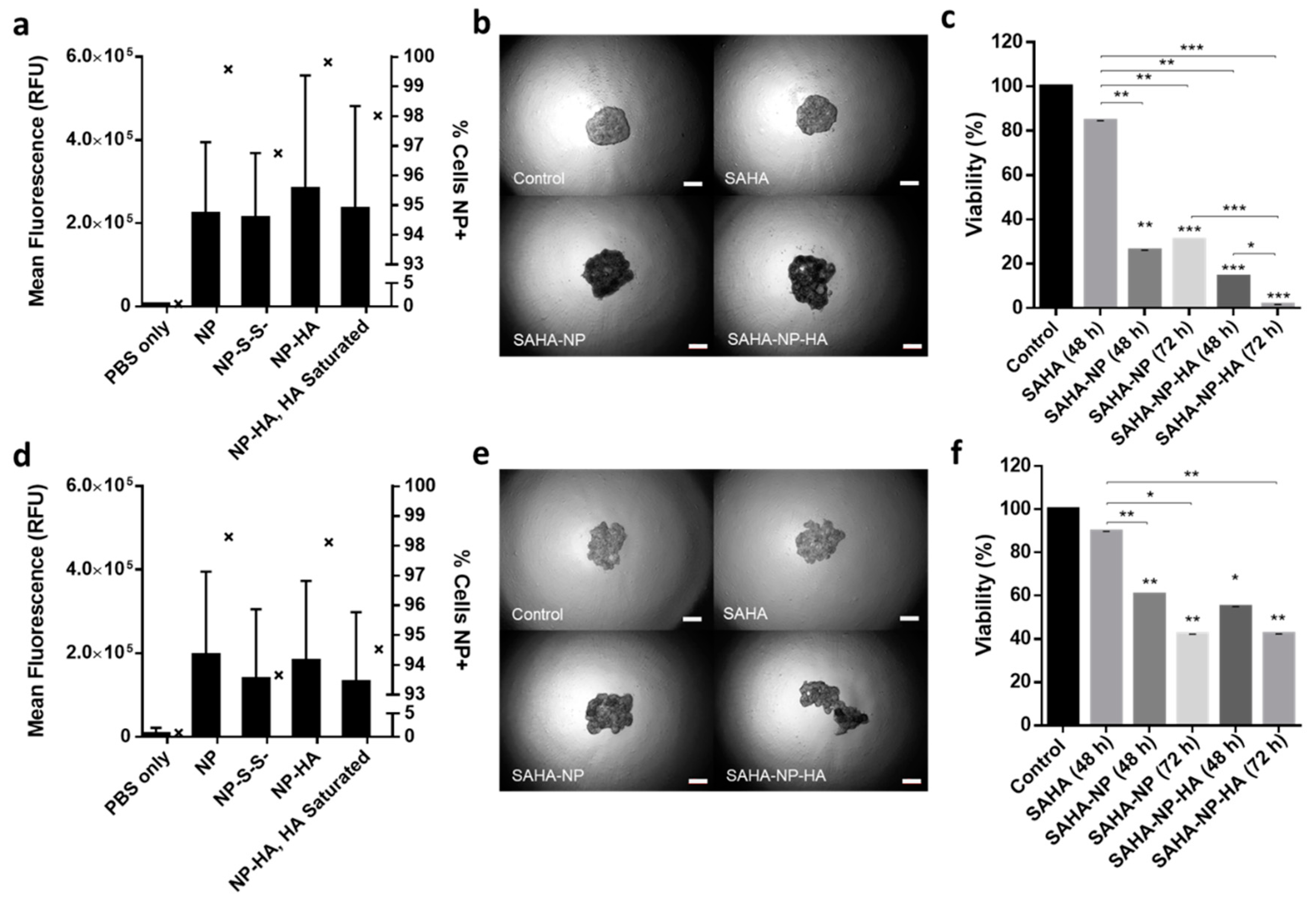
3.3. NP Cellular Uptake and Biocompatibility
3.4. Micelle Encapsulation Enhances SAHA Cytotoxicity
3.5. SAHA-NP Treatment Alters Histone H3 Acetylation at the Promoters of p21 and p53
3.6. HA Enhances Cytotoxicity of SAHA Nanoparticles in 3D Tumor Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lortet-Tieulent, J.; Ferlay, J.; Bray, F.; Jemal, A. International patterns and trends in endometrial cancer incidence, 1978–2013. J. Natl. Cancer Inst. 2018, 110, 354–361. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Intelligence Network (NCIN) Outline of Uterine Cancer in the United Kingdom: Incidence, Mortality and Survival. Available online: http://www.ncin.org.uk/view?rid=2398#:~:text=In%20the%20UK%2C%20the%20incidence,3.2%20to%203.7%20per%20100%2C000 (accessed on 15 August 2013).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Cancer Research UK. Uterine Cancer Incidence; Cancer Research: London, UK, 2021. [Google Scholar]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Saso, S.; Chatterjee, J.; Georgiou, E.; Ditri, A.M.; Smith, J.R.; Ghaem-Maghami, S. Endometrial cancer. BMJ 2011, 343. [Google Scholar] [CrossRef] [Green Version]
- Frei, K.A.; Kinkel, K. Staging endometrial cancer: Role of magnetic resonance imaging. J. Magn. Reson. Imaging 2001, 13, 850–855. [Google Scholar] [CrossRef]
- Doll, A.; Abal, M.; Rigau, M.; Monge, M.; Gonzalez, M.; Demajo, S.; Colás, E.; Llauradó, M.; Alazzouzi, H.; Planagumá, J.; et al. Novel molecular profiles of endometrial cancer-new light through old windows. J. Steroid Biochem. Mol. Biol. 2008, 108, 221–229. [Google Scholar] [CrossRef]
- Hoffman, B.L.; Schorge, J.O.; Schaffer, J.I.; Halvorson, L.M.; Bradshaw, K.D.; Cunningham, F.G.; Calver, L.E. Chapter 15. Reproductive Endocrinology; Williams Gynecology: New York, NY, USA, 2012. [Google Scholar]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. DNA Damage and Repair and Their Role in Carcinogenesis. In Molecular Cell Biology, 4th ed.; Freeman, W.H., Ed.; Macmillan: New York, NY, USA, 2000. [Google Scholar]
- Vale, C.L.; Tierney, J.; Bull, S.J.; Symonds, P.R. Chemotherapy for advanced, recurrent or metastatic endometrial carcinoma. Cochrane Database Syst. Rev. 2012, 8, CD003915. [Google Scholar] [CrossRef] [PubMed]
- Risinger, J.I.; Maxwell, G.L.; Chandramouli, G.V.R.; Jazaeri, A.; Aprelikova, O.; Patterson, T.; Berchuck, A.; Barrett, J.C. Microarray analysis reveals distinct gene expression profiles among different histologic types of endometrial cancer. Cancer Res. 2003, 63, 6–11. [Google Scholar] [PubMed]
- Nagashima, M.; Miwa, N.; Hirasawa, H.; Katagiri, Y.; Takamatsu, K.; Morita, M. Genome-wide DNA methylation analysis in obese women predicts an epigenetic signature for future endometrial cancer. Sci. Rep. 2019, 9, 6469. [Google Scholar] [CrossRef] [Green Version]
- Tao, M.H.; Freudenheim, J.L. DNA methylation in endometrial cancer. Epigenetics 2010, 5, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Sakuragi, N. Recent advances in research on epigenetic alterations and clinical significance of para-aortic lymphadenectomy in endometrial cancer: An introduction. Int. J. Clin. Oncol. 2013, 18, 183–185. [Google Scholar] [CrossRef]
- Arafa, M.; Somja, J.; Dehan, P.; Kridelka, F.; Goffin, F.; Boniver, J.; Delvenne, P. Current concepts in the pathology and epigenetics of endometrial carcinoma. Pathology 2010, 42, 613–617. [Google Scholar] [CrossRef]
- Bartosch, C.; Lopes, J.M.; Jerónimo, C. Epigenetics in endometrial carcinogenesis—Part 1: DNA methylation. Epigenomics 2017, 9, 737–755. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.D.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.; Boshoff, C.; Beck, S. Cancer Epigenome. Adv. Genet. 2010, 70, 247–276. [Google Scholar] [PubMed]
- Ma, X.; Ma, C.X.; Wang, J. Endometrial carcinogenesis and molecular signaling pathways. Am. J. Mol. Biol. 2014, 4, 134–149. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Zhang, J.; Cai, H.; Li, Y.; Zhang, Y.; Zhang, X.; Zhao, D.; Li, Z.; Ma, H.; Wang, J.; et al. HDAC as a therapeutic target for treatment of endometrial cancers. Curr. Pharm. Des. 2014, 20, 1847–1856. [Google Scholar] [CrossRef]
- Jiang, S.; Dowdy, S.C.; Meng, X.W.; Wang, Z.; Jones, M.B.; Podratz, K.C.; Jiang, S.W. Histone deacetylase inhibitors induce apoptosis in both Type I and Type II endometrial cancer cells. Gynecol. Oncol. 2007, 105, 493–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, N.; Narahara, H.; Takai, N.; Narahara, H. Human endometrial and ovarian cancer cells: Histone deacetylase inhibitors exhibit antiproliferative activity, potently induce cell cycle arrest, and stimulate apoptosis. Curr. Med. Chem. 2007, 14, 2548–2553. [Google Scholar] [CrossRef]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Zatloukal, K. Molecular mechanism leading to SAHA-induced autophagy in tumor cells: Evidence for a p53-dependent pathway. Cancer Cell Int. 2016, 16, 68. [Google Scholar] [CrossRef] [Green Version]
- Nervi, C.; De Marinis, E.; Codacci-Pisanelli, G. Epigenetic treatment of solid tumours: A review of clinical trials. Clin. Epigenet. 2015, 7, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, H.; Watanabe, R.; Choyke, P.L. Improving conventional enhanced permeability and retention (EPR) effects; What is the appropriate target? Theranostics 2014, 4, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Jia, L. Nanoparticle formulation increases oral bioavailability of poorly soluble drugs: Approaches, experimental evidences and theory. Curr. Nanosci. 2005, 1, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Ebeid, K.; Meng, X.; Thiel, K.W.; Do, A.V.; Geary, S.M.; Morris, A.S.; Pham, E.L.; Wongrakpanich, A.; Chhonker, Y.S.; Murry, D.J.; et al. Synthetically lethal nanoparticles for treatment of endometrial cancer. Nat. Nanotechnol. 2018, 13, 72–81. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Liu, L.; Xue, X.; Liang, X.-J. Nanoparticle-based drug delivery systems: What can they really do in vivo? F1000Research 2017, 6, 681. [Google Scholar] [CrossRef]
- Safra, T.; Muggia, F.; Jeffers, S.; Tsao-Wei, D.D.; Groshen, S.; Lyass, O.; Henderson, R.; Berry, G.; Gabizon, A. Pegylated liposomal doxorubicin (doxil): Reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann. Oncol. 2000, 11, 1029–1033. [Google Scholar] [CrossRef]
- Clift, A.K.; Drymousis, P.; Al-Nahhas, A.; Wasan, H.; Martin, J.; Holm, S.; Frilling, A. Incidence of second primary malignancies in patients with neuroendocrine tumours. Neuroendocrinology 2015, 102, 26–32. [Google Scholar] [CrossRef]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Rompicharla, S.V.K.; Trivedi, P.; Kumari, P.; Ghanta, P.; Ghosh, B.; Biswas, S. Polymeric micelles of suberoylanilide hydroxamic acid to enhance the anticancer potential in vitro and in vivo. Nanomedicine 2017, 12, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sun, J.; Wang, P.; Ma, X.; Li, S. Pendant HDAC inhibitor SAHA derivatised polymer as a novel prodrug micellar carrier for anticancer drugs. J. Drug Target. 2018, 26, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Mattheolabakis, G.; Milane, L.; Singh, A.; Amiji, M.M. Hyaluronic acid targeting of CD44 for cancer therapy: From receptor biology to nanomedicine. J. Drug Target. 2015, 23, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Choi, J.Y.; Ramasamy, T.; Truong, D.H.; Nguyen, C.N.; Choi, H.G.; Yong, C.S.; Kim, J.O. Hyaluronic acid-coated solid lipid nanoparticles for targeted delivery of vorinostat to CD44 overexpressing cancer cells. Carbohydr. Polym. 2014, 114, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: Applications, advantages and disadvantages. Res. Pharm. Sci. 2018, 13, 288–303. [Google Scholar]
- Khatak, S.; Dureja, H. Recent techniques and patents on solid lipid nanoparticles as novel carrier for drug delivery. Recent Pat. Nanotechnol. 2015, 9, 150–177. [Google Scholar] [CrossRef]
- Jahangirian, H.; Lemraski, E.G.; Webster, T.J.; Rafiee-Moghaddam, R.; Abdollahi, Y. A review of drug delivery systems based on nanotechnology and green chemistry: Green nanomedicine. Int. J. Nanomed. 2017, 12, 2957–2978. [Google Scholar] [CrossRef] [Green Version]
- De Jong, W.H.; Borm, P.J.A. Drug delivery and nanoparticles: Applications and hazards. Int. J. Nanomed. 2008, 3, 133–149. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.H.; Park, K.; Han, D.K. Preparation of TGF-β1-conjugated biodegradable pluronic F127 hydrogel and its application with adipose-derived stem cells. J. Control Release 2010, 147, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Akash, M.S.H.; Rehman, K.; Chen, S. Pluronic F127-based thermosensitive gels for delivery of therapeutic proteins and peptides. Polym. Rev. 2014, 54, 573–597. [Google Scholar] [CrossRef]
- Diniz, I.M.A.; Chen, C.; Xu, X.; Ansari, S.; Zadeh, H.H.; Marques, M.M.; Shi, S.; Moshaverinia, A. Pluronic F-127 hydrogel as a promising scaffold for encapsulation of dental-derived mesenchymal stem cells. J. Mater. Sci. Mater. Med. 2015, 26, 153. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, S.; Yokouchi, C.; Nakamura, T.; Hashiguchi, N.; Hou, W.-M.; Takada, M. Pluronic F-127 gels as a novel vehicle for rectal administration of indomethacin. Chem. Pharm. Bull. 1986, 34, 1801–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Williams, G.R.; Wu, J.; Wu, J.; Niu, S.; Xie, X.; Li, S.; Zhu, L.M. Pluronic F127-based micelles for tumor-targeted bufalin delivery. Int. J. Pharm. 2019, 559, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Shi, H.; Qiao, M.; Zhang, Z.; Yang, W.; Dong, L.; Xie, F.; Zhao, C.; Kang, L. PH-sensitive micelles for the intracellular co-delivery of curcumin and Pluronic L61 unimers for synergistic reversal effect of multidrug resistance. Sci. Rep. 2017, 7, 42465. [Google Scholar] [CrossRef] [Green Version]
- Misra, S.; Heldin, P.; Hascall, V.C.; Karamanos, N.K.; Skandalis, S.S.; Markwald, R.R.; Ghatak, S. Hyaluronan-CD44 interactions as potential targets for cancer therapy. FEBS J. 2011, 278, 1429–1443. [Google Scholar] [CrossRef] [Green Version]
- Afify, A.M.; Craig, S.; Paulino, A.F.G.; Stern, R. Expression of hyaluronic acid and its receptors, CD44s and CD44v6, in normal, hyperplastic, and neoplastic endometrium. Ann. Diagn. Pathol. 2005, 9, 312–318. [Google Scholar] [CrossRef]
- Wojciechowski, M.; Krawczyk, T.; Śmigielski, J.; Malinowski, A. CD44 expression in curettage and postoperative specimens of endometrial cancer. Arch. Gynecol. Obstet. 2015, 291, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Pulakkat, S.; Balaji, S.A.; Rangarajan, A.; Raichur, A.M. Surface Engineered Protein Nanoparticles with Hyaluronic Acid Based Multilayers for Targeted Delivery of Anticancer Agents. ACS Appl. Mater. Interfaces 2016, 8, 23437–23449. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Min, K.H.; Na, J.H.; Choi, K.; Kim, K.; Park, J.H.; Kwon, I.C.; Jeong, S.Y. Self-assembled hyaluronic acid nanoparticles as a potential drug carrier for cancer therapy: Synthesis, characterization, and in vivo biodistribution. J. Mater. Chem. 2009, 19, 4102–4107. [Google Scholar] [CrossRef]
- Kim, M.; Hwang, Y.; Tae, G. The enhanced anti-tissue adhesive effect of injectable pluronic-HA hydrogel by poly(γ-glutamic acid). Int. J. Biol. Macromol. 2016, 93, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Wu, H.C.; Sun, J.S.; Dong, G.C.; Wang, T.W. Injectable and thermoresponsive self-assembled nanocomposite hydrogel for long-term anticancer drug delivery. Langmuir 2013, 29, 3721–3729. [Google Scholar] [CrossRef]
- Kozak, J.; Wdowiak, P.; Maciejewski, R.; Torres, A. A guide for endometrial cancer cell lines functional assays using the measurements of electronic impedance. Cytotechnology 2018, 70, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, R.L.F.; Miranda, M.; Marcato, P.D.; Swiech, K. Comparative analysis of 3D bladder tumor spheroids obtained by forced floating and hanging drop methods for drug screening. Front. Physiol. 2017, 8, 605. [Google Scholar] [CrossRef] [Green Version]
- Costa, E.C.; Diogo, D.M.D.M.; Moreira, A.F.; Carvalho, M.P.; Correia, I.J. Spheroids formation on non-adhesive surfaces by liquid overlay technique: Considerations and practical approaches. Biotechnol. J. 2018, 13, 1700417. [Google Scholar] [CrossRef]
- Gurav, D.; Varghese, O.P.; Hamad, O.A.; Nilsson, B.; Hilborn, J.; Oommen, O.P. Chondroitin sulfate coated gold nanoparticles: A new strategy to resolve multidrug resistance and thromboinflammation. Chem. Commun. 2016, 52, 966–969. [Google Scholar] [CrossRef]
- Koivusalo, L.; Kauppila, M.; Samanta, S.; Parihar, V.S.; Ilmarinen, T.; Miettinen, S.; Oommen, O.P.; Skottman, H. Tissue adhesive hyaluronic acid hydrogels for sutureless stem cell delivery and regeneration of corneal epithelium and stroma. Biomaterials 2019, 225, 119516. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, K.M.E.; Carlsson, P.J.E.; Li, J.T. Coating of Hydrophobic Surfaces to Render them Protein Resistant while Permitting Covalent Attachment of Specific Ligands; Patent and Trademark Office: Washington, DC, USA, 1998. [Google Scholar]
- Li, J.-T.; Carlsson, J.; Lin, J.-N.; Caldwell, K.D. Chemical modification of surface active Poly(ethylene oxide)−Poly(propylene oxide) triblock copolymers. Bioconjug. Chem. 1996, 7, 592–599. [Google Scholar] [CrossRef]
- Horcas, I.; Fernández, R.; Gómez-Rodríguez, J.M.; Colchero, J.; Gómez-Herrero, J.; Baro, A.M. WSXM: A software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 2007, 78, 013705. [Google Scholar] [CrossRef]
- Kwak, T.W.; Kim, D.H.; Jeong, Y.-I.; Kang, D.H. Antitumor activity of vorinostat-incorporated nanoparticles against human cholangiocarcinoma cells. J. Nanobiotechnol. 2015, 13, 60. [Google Scholar] [CrossRef] [Green Version]
- Holmes, K.A.; Brown, G.D.; Carroll, J.S. Chromatin immunoprecipitation-sequencing (ChLP-seq) for mapping of estrogen receptor-chromatin interactions in breast cancer. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; pp. 79–98. [Google Scholar]
- Hunter, A.L.; Narang, N.; Baxter, M.; Ray, D.W.; Poolman, T.M. An improved method for quantitative ChIP studies of nuclear receptor function. J. Mol. Endocrinol. 2019, 62, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Idrees, A.; Chiono, V.; Ciardelli, G.; Shah, S.; Viebahn, R.; Zhang, X.; Salber, J. Validation of in vitro assays in three-dimensional human dermal constructs. Int. J. Artif. Organs 2018, 41, 779–788. [Google Scholar] [CrossRef]
- Duellman, S.J.; Zhou, W.; Meisenheimer, P.; Vidugiris, G.; Cali, J.J.; Gautam, P.; Wennerberg, K.; Vidugiriene, J. Bioluminescent, nonlytic, real-time cell viability assay and use in inhibitor screening. Assay Drug Dev. Technol. 2015, 13, 456–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, W.K.; Marks, P.A. Drug insight: Histone deacetylase inhibitors—Development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat. Clin. Pr. Oncol. 2005, 2, 150–157. [Google Scholar] [CrossRef]
- Komatsu, N.; Kawamata, N.; Takeuchi, S.; Yin, D.; Chien, W.; Miller, C.W.; Koeffler, H.P. SAHA, a HDAC inhibitor, has profound anti-growth activity against non-small cell lung cancer cells. Oncol. Rep. 2006, 15, 187–191. [Google Scholar] [CrossRef]
- Soderberg, T. 15.7: Redox reactions of thiols and disulfides. In Organic Chemistry with a Biological Emphasis Volume I; Chemistry Publications; University of Minnesota Morris Digital Well: Morris, MI, USA, 2019; Volume 1, Available online: https://digitalcommons.morris.umn.edu/chem_facpubs/1/ (accessed on 7 July 2021).
- Edwardson, J.M.; Henderson, R.M. Atomic force microscopy and drug discovery. Drug Discov. Today 2004, 9, 64–71. [Google Scholar] [CrossRef]
- Maver, U.; Velnar, T.; Gaberšček, M.; Planinšek, O.; Finšgar, M. Recent progressive use of atomic force microscopy in biomedical applications. TrAC Trends Anal. Chem. 2016, 80, 96–111. [Google Scholar] [CrossRef] [Green Version]
- Atomic Force Microscopy in Biomedical Research; Humana Press: New York, NY, USA, 2011; ISBN 978-1-61779-104-8.
- Eaton, P.; Quaresma, P.; Soares, C.; Neves, C.; De Almeida, M.P.; Pereira, E.; West, P. A direct comparison of experimental methods to measure dimensions of synthetic nanoparticles. Ultramicroscopy 2017, 182, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Pisano, S.; Wang, X.; Garcia-Parra, J.; Gazze, A.; Edwards, K.; Feltracco, V.; Hu, Y.; He, L.; Gonzalez, D.; Francis, L.W.; et al. Nanomicelles potentiate histone deacetylase inhibitor efficacy in vitro. Cancer Nanotechnol. 2020, 11, 14. [Google Scholar] [CrossRef]
- Teulon, J.M.; Godon, C.; Chantalat, L.; Moriscot, C.; Cambedouzou, J.; Odorico, M.; Ravaux, J.; Podor, R.; Gerdil, A.; Habert, A.; et al. On the operational aspects of measuring nanoparticle sizes. Nanomaterials 2019, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Gref, R.; Torchilin, V.; Minamitake, Y.; Peracchia, M.; Trubetskoy, V.; Langer, R. Biodegradable long-circulating polymeric nanospheres. Science 2006, 263, 1600–1603. [Google Scholar] [CrossRef] [Green Version]
- Dinarvand, R.; Sepehri, N.; Manoochehri, S.; Rouhani, H.; Atyabi, F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int. J. Nanomed. 2011, 6, 877–895. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Nie, X.; Zou, M.; Shi, Y.; Cheng, G. Recent advances in materials for extended-release antibiotic delivery system. J. Antibiot. 2011, 64, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Stiefel, P.; Schmidt-Emrich, S.; Maniura-Weber, K.; Ren, Q. Critical aspects of using bacterial cell viability assays with the fluorophores SYTO9 and propidium iodide. BMC Microbiol. 2015, 15, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes-Alves, A.G.; Maia, A.F.; Cruz, T.; Castro, H.; Tomás, A.M. Development of an automated image analysis protocol for quantification of intracellular forms of Leishmania spp. PLoS ONE 2018, 13, e0201747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nykopp, T.K.; Rilla, K.; Tammi, M.I.; Tammi, R.H.; Sironen, R.; Hämäläinen, K.; Kosma, V.M.; Heinonen, S.; Anttila, M. Hyaluronan synthases (HAS1-3) and hyaluronidases (HYAL1-2) in the accumulation of hyaluronan in endometrioid endometrial carcinoma. BMC Cancer 2010, 10, 512. [Google Scholar] [CrossRef] [Green Version]
- Nykopp, T.K.; Pasonen-Seppänen, S.; Tammi, M.I.; Tammi, R.H.; Kosma, V.M.; Anttila, M.; Sironen, R. Decreased hyaluronidase 1 expression is associated with early disease recurrence in human endometrial cancer. Gynecol. Oncol. 2015, 137, 152–159. [Google Scholar] [CrossRef]
- Paiva, P.; Van Damme, M.P.; Tellbach, M.; Jones, R.L.; Jobling, T.; Salamonsen, L.A. Expression patterns of hyaluronan, hyaluronan synthases and hyaluronidases indicate a role for hyaluronan in the progression of endometrial cancer. Gynecol. Oncol. 2005, 98, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, H.; Maruyama, T.; Nishikawa-Uchida, S.; Oda, H.; Miyazaki, K.; Yamasaki, A.; Yoshimura, Y. Studies using an in vitro model show evidence of involvement of epithelial-mesenchymal transition of human endometrial epithelial cells in human embryo implantation. J. Biol. Chem. 2012, 287, 4441–4450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Terai, Y.; Kawaguchi, H.; Fujiwara, S.; Yoo, S.; Tsunetoh, S.; Takai, M.; Kanemura, M.; Tanabe, A.; Ohmichi, M. Prognostic impact of EMT (epithelial-mesenchymal-transition)-related protein expression in endometrial cancer. Cancer Biol. Ther. 2013, 14, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.Y.; Chen, C.S.; Lin, S.P.; Weng, J.R.; Chen, C.S. Targeting histone deacetylase in cancer therapy. Med. Res. Rev. 2006, 26, 397–413. [Google Scholar] [CrossRef]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Lovitt, C.; Shelper, T.; Avery, V. Advanced cell culture techniques for cancer drug discovery. Biology 2014, 3, 345. [Google Scholar] [CrossRef] [Green Version]
- Nirmalanandhan, V.S.; Duren, A.; Hendricks, P.; Vielhauer, G.; Sittampalam, G.S. Activity of anticancer agents in a three-dimensional cell culture model. Assay Drug Dev. Technol. 2010, 8, 581–590. [Google Scholar] [CrossRef]
- Lagies, S.; Schlimpert, M.; Neumann, S.; Wäldin, A.; Kammerer, B.; Borner, C.; Peintner, L. Cells grown in three-dimensional spheroids mirror in vivo metabolic response of epithelial cells. Commun. Biol. 2020, 3, 246. [Google Scholar] [CrossRef]
- Chitcholtan, K.; Asselin, E.; Parent, S.; Sykes, P.H.; Evans, J.J. Differences in growth properties of endometrial cancer in three dimensional (3D) culture and 2D cell monolayer. Exp. Cell Res. 2013, 319, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, N.; Zhou, W.; Xu, J.; Chen, J.; Zheng, F.; Long, Z.; Yue, C.; Ai, K.; Liu, L.; et al. Up-regulation of P21 inhibits TRAIL-mediated extrinsic apoptosis, contributing resistance to SAHA in acute myeloid leukemia cells. Cell. Physiol. Biochem. 2014, 34, 506–518. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Z.; Wang, H.; Liu, X.; Zhou, Z.; Tang, J.; Liu, X.; Zheng, M.; Shen, Y. SAHA (vorinostat) facilitates functional polymer-based gene transfection via upregulation of ROS and synergizes with TRAIL gene delivery for cancer therapy. J. Drug Target. 2019, 27, 306–314. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle | Size Average (nm) | PDI | Zeta Potential (mV) | Encapsulation Efficiency (EE%) | Loading Capacity (LC%) | Drug Release Maxima (h) | Encapsulated Drug Released over 72 h | First Order Rate Constant |
|---|---|---|---|---|---|---|---|---|
| Empty NP | 29.1 ± 2.1 | 0.243 | 0.120 ± 0.25 | - | - | - | - | - |
| SAHA-NP | 35.7 ± 3.6 | 0.354 | −0.197 ± 0.15 | 18% | 0.6% | 2 | 28.06 µg | 2.5 ± 0.2 |
| SAHA-NP-S-S- | 179.9 ± 15.0 | 0.229 | −3.12 ± 0.61 | 42% | 9.1% | 0.5 | 44.61 µg | - |
| SAHA-NP-HA | 251.6 ± 22.0 | 0.312 | −14.23 ± 3.29 | 45% ** | 9.7% ** | 23.5 | 39.63 µg | 0.3 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edwards, K.; Yao, S.; Pisano, S.; Feltracco, V.; Brusehafer, K.; Samanta, S.; Oommen, O.P.; Gazze, S.A.; Paravati, R.; Maddison, H.; et al. Hyaluronic Acid-Functionalized Nanomicelles Enhance SAHA Efficacy in 3D Endometrial Cancer Models. Cancers 2021, 13, 4032. https://doi.org/10.3390/cancers13164032
Edwards K, Yao S, Pisano S, Feltracco V, Brusehafer K, Samanta S, Oommen OP, Gazze SA, Paravati R, Maddison H, et al. Hyaluronic Acid-Functionalized Nanomicelles Enhance SAHA Efficacy in 3D Endometrial Cancer Models. Cancers. 2021; 13(16):4032. https://doi.org/10.3390/cancers13164032
Chicago/Turabian StyleEdwards, Kadie, Seydou Yao, Simone Pisano, Veronica Feltracco, Katja Brusehafer, Sumanta Samanta, Oommen P. Oommen, S. Andrea Gazze, Roberta Paravati, Holly Maddison, and et al. 2021. "Hyaluronic Acid-Functionalized Nanomicelles Enhance SAHA Efficacy in 3D Endometrial Cancer Models" Cancers 13, no. 16: 4032. https://doi.org/10.3390/cancers13164032
APA StyleEdwards, K., Yao, S., Pisano, S., Feltracco, V., Brusehafer, K., Samanta, S., Oommen, O. P., Gazze, S. A., Paravati, R., Maddison, H., Li, C., Gonzalez, D., Conlan, R. S., & Francis, L. (2021). Hyaluronic Acid-Functionalized Nanomicelles Enhance SAHA Efficacy in 3D Endometrial Cancer Models. Cancers, 13(16), 4032. https://doi.org/10.3390/cancers13164032