
Targetable Molecular Alterations in the Treatment of Biliary Tract Cancers: An Overview of the Available Treatments
 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
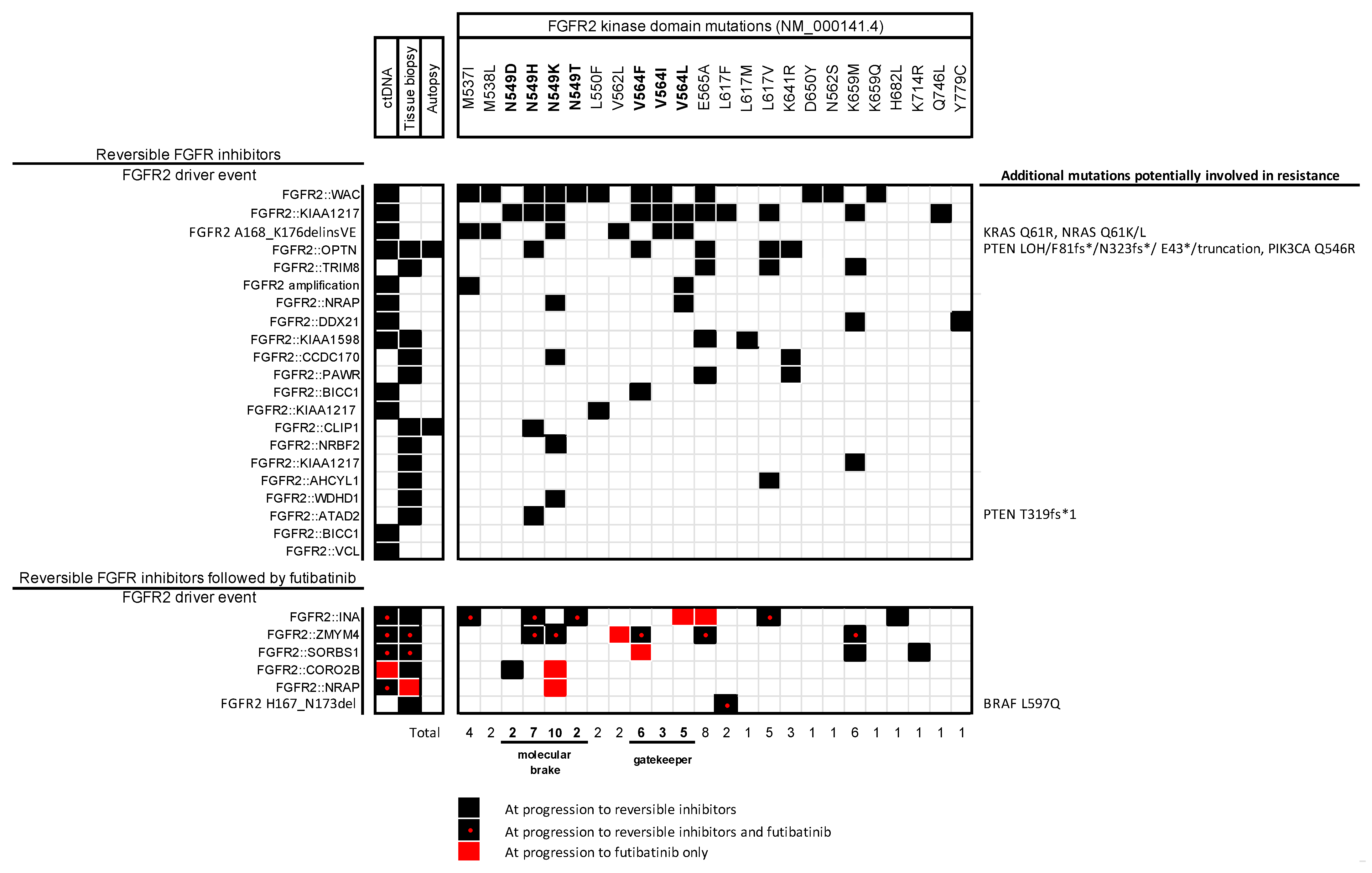
1.1. FGFR Fusions
1.2. NTRK Fusions
1.3. Isocitrate Dehydrogenase 1 (IDH1) Mutation
1.4. HER2 Mutations and Amplifications
1.5. KRAS G12C Mutation
1.6. BRAF Mutation
1.7. Mutation of Genes Involved in DNA Repair
1.7.1. Mismatch Repair System (MMR)
1.7.2. BRCA1/2 Mutations
1.7.3. Other Genes Involved in DNA Repair: ARID1A, BAP1, ATM and ATR
1.8. Molecular Profile: New Approaches to Guide the Treatment of BTC?
2. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bridgewater, J.A.; Goodman, K.A.; Kalyan, A.; Mulcahy, M.F. Biliary Tract Cancer: Epidemiology, Radiotherapy, and Molecular Profiling. Am. Soc. Clin. Oncol. Educ. Book 2016, 36, e194–e203. [Google Scholar] [CrossRef]
- Vogel, A.; Bridgewater, J.; Edeline, J.; Kelley, R.K.; Klümpen, H.J.; Malka, D.; Primrose, J.N.; Rimassa, L.; Stenzinger, A.; Valle, J.W.; et al. Biliary Tract Cancer: ESMO Clinical Practice Guideline for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2023, 34, 127–140. [Google Scholar] [CrossRef]
- Malka, D.; Blanc, J.F.; Boudjema, K.; Bretagne-Bignon, A.L.; Coriat, R.; de Baère, T.; de la Fouchardière, C.; Dromain, C.; Edeline, J.; Gelli, M.; et al. « Cancer Des Voies Biliaires ». Thésaurus National de Cancérologie Digestive, Juillet 2022. Available online: https://www.snfge.org/content/8-cancer-des-voies-biliaires (accessed on 28 August 2023).
- Oh, D.-Y.; Ruth, H.A.; Qin, S.; Chen, L.-T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Ah, L.M.; Kitano, M.; et al. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer. NEJM Evid. 2022, 1, EVIDoa2200015. [Google Scholar] [CrossRef]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. Second-Line FOLFOX Chemotherapy versus Active Symptom Control for Advanced Biliary Tract Cancer (ABC-06): A Phase 3, Open-Label, Randomised, Controlled Trial. Lancet Oncol. 2021, 22, 690–701. [Google Scholar] [CrossRef]
- Kelley, R.K.; Ueno, M.; Yoo, C.; Finn, R.S.; Furuse, J.; Ren, Z.; Yau, T.; Klümpen, H.-J.; Chan, S.L.; Ozaka, M.; et al. Pembrolizumab in Combination with Gemcitabine and Cisplatin Compared with Gemcitabine and Cisplatin Alone for Patients with Advanced Biliary Tract Cancer (KEYNOTE-966): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2023, 401, 1853–1865. [Google Scholar] [CrossRef]
- Valle, J.W.; Borbath, I.; Khan, S.A.; Huguet, F.; Gruenberger, T.; Arnold, D. Biliary Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2016, 27, v28–v37. [Google Scholar] [CrossRef]
- Lamarca, A.; Kapacee, Z.; Breeze, M.; Bell, C.; Belcher, D.; Staiger, H.; Taylor, C.; McNamara, M.G.; Hubner, R.A.; Valle, J.W. Molecular Profiling in Daily Clinical Practice: Practicalities in Advanced Cholangiocarcinoma and Other Biliary Tract Cancers. J. Clin. Med. 2020, 9, 2854. [Google Scholar] [CrossRef]
- Berchuck, J.E.; Facchinetti, F.; DiToro, D.F.; Baiev, I.; Majeed, U.; Reyes, S.; Chen, C.; Zhang, K.; Sharman, R.; Uson Junior, P.L.S.; et al. The Clinical Landscape of Cell-Free DNA Alterations in 1671 Patients with Advanced Biliary Tract Cancer. Ann. Oncol. 2022, 33, 1269–1283. [Google Scholar] [CrossRef]
- Javle, M.; Bekaii-Saab, T.; Jain, A.; Wang, Y.; Kelley, R.K.; Wang, K.; Kang, H.C.; Catenacci, D.; Ali, S.; Krishnan, S.; et al. Biliary Cancer: Utility of next-Generation Sequencing for Clinical Management. Cancer 2016, 122, 3838–3847. [Google Scholar] [CrossRef]
- Verlingue, L.; Malka, D.; Allorant, A.; Massard, C.; Ferté, C.; Lacroix, L.; Rouleau, E.; Auger, N.; Ngo, M.; Nicotra, C.; et al. Precision Medicine for Patients with Advanced Biliary Tract Cancers: An Effective Strategy within the Prospective MOSCATO-01 Trial. Eur. J. Cancer 2017, 87, 122–130. [Google Scholar] [CrossRef]
- Ong, C.K.; Subimerb, C.; Pairojkul, C.; Wongkham, S.; Cutcutache, I.; Yu, W.; McPherson, J.R.; Allen, G.E.; Ng, C.C.Y.; Wong, B.H.; et al. Exome Sequencing of Liver Fluke-Associated Cholangiocarcinoma. Nat. Genet. 2012, 44, 690–693. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for Previously Treated, Locally Advanced or Metastatic Cholangiocarcinoma: A Multicentre, Open-Label, Phase 2 Study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-Mutant, Chemotherapy-Refractory Cholangiocarcinoma (ClarIDHy): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.K.Y.; Bedard, P.L.; Tortora, G.; Douillard, J.-Y.; et al. A Framework to Rank Genomic Alterations as Targets for Cancer Precision Medicine: The ESMO Scale for Clinical Actionability of Molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.B.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the Use of Next-Generation Sequencing (NGS) for Patients with Metastatic Cancers: A Report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Silverman, I.M.; Hollebecque, A.; Friboulet, L.; Owens, S.; Newton, R.C.; Zhen, H.; Féliz, L.; Zecchetto, C.; Melisi, D.; Burn, T.C. Clinicogenomic Analysis of FGFR2-Rearranged Cholangiocarcinoma Identifies Correlates of Response and Mechanisms of Resistance to Pemigatinib. Cancer Discov. 2021, 11, 326–339. [Google Scholar] [CrossRef]
- Benayed, R.; Offin, M.; Mullaney, K.; Sukhadia, P.; Rios, K.; Desmeules, P.; Ptashkin, R.; Won, H.; Chang, J.; Halpenny, D.; et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin. Cancer Res. 2019, 25, 4712–4722. [Google Scholar] [CrossRef]
- De Luca, A.; Esposito Abate, R.; Rachiglio, A.M.; Maiello, M.R.; Esposito, C.; Schettino, C.; Izzo, F.; Nasti, G.; Normanno, N. FGFR Fusions in Cancer: From Diagnostic Approaches to Therapeutic Intervention. Int. J. Mol. Sci. 2020, 21, 6856. [Google Scholar] [CrossRef]
- Evers, D.L.; He, J.; Kim, Y.H.; Mason, J.T.; O’Leary, T.J. Paraffin Embedding Contributes to RNA Aggregation, Reduced RNA Yield, and Low RNA Quality. J. Mol. Diagn. 2011, 13, 687–694. [Google Scholar] [CrossRef]
- Pereira, B.; Chen, C.T.; Goyal, L.; Walmsley, C.; Pinto, C.J.; Baiev, I.; Allen, R.; Henderson, L.; Saha, S.; Reyes, S.; et al. Cell-Free DNA Captures Tumor Heterogeneity and Driver Alterations in Rapid Autopsies with Pre-Treated Metastatic Cancer. Nat. Commun. 2021, 12, 3199. [Google Scholar] [CrossRef]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Russano, M.; Napolitano, A.; Ribelli, G.; Iuliani, M.; Simonetti, S.; Citarella, F.; Pantano, F.; Dell’Aquila, E.; Anesi, C.; Silvestris, N.; et al. Liquid Biopsy and Tumor Heterogeneity in Metastatic Solid Tumors: The Potentiality of Blood Samples. J. Exp. Clin. Cancer Res. 2020, 39, 95. [Google Scholar] [CrossRef] [PubMed]
- Mondaca, S.; Nervi, B.; Pinto, M.; Abou-Alfa, G.K. Biliary Tract Cancer Prognostic and Predictive Genomics. Chin. Clin. Oncol. 2019, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Koshiol, J.; Yu, B.; Kabadi, S.M.; Baria, K.; Shroff, R.T. Epidemiologic Patterns of Biliary Tract Cancer in the United States: 2001–2015. BMC Cancer 2022, 22, 1178. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Hu, J.; Liu, S.; Meric-Bernstam, F.; Abdel-Wahab, R.; Xu, J.; Li, Q.; Yan, M.; Feng, Y.; Lin, J.; et al. Intrahepatic Cholangiocarcinoma: Genomic Heterogeneity Between Eastern and Western Patients. JCO Precis. Oncol. 2020, 4, PO.18.00414. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xu, Y.; Gao, W.; Li, M.; He, J.; Deng, X.; Xing, W. Comprehensive Germline and Somatic Genomic Profiles of Chinese Patients with Biliary Tract Cancer. Front. Oncol. 2022, 12, 930611. [Google Scholar] [CrossRef] [PubMed]
- Antoun, L.; Deneche, I.; Boileve, A.; Tarabay, A.; Rouleau, E.; El Rawadi, E.; Eid, R.; Lacroix, L.; Valéry, M.; Boige, V.; et al. SO-4 Impact of molecular profiling on survival in patients with advanced biliary tract cancers. Ann. Oncol. 2023, 34, S163. [Google Scholar] [CrossRef]
- Katoh, M. Fibroblast Growth Factor Receptors as Treatment Targets in Clinical Oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next Horizon in Mechanisms and Management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic Spectra of Biliary Tract Cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast Growth Factor Receptor 2 Tyrosine Kinase Fusions Define a Unique Molecular Subtype of Cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.M.; Murugesan, K.; Shroff, R.T.; Borad, M.J.; Abdel-Wahab, R.; Schrock, A.B.; Chung, J.; Goyal, L.; Frampton, G.M.; Kelley, R.K.; et al. Profiling of 3,634 Cholangiocarcinomas (CCA) to Identify Genomic Alterations (GA), Tumor Mutational Burden (TMB), and Genomic Loss of Heterozygosity (GLOH). J. Clin. Oncol. 2019, 37, 4087. [Google Scholar] [CrossRef]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Molecular Targeted Therapies: Ready for “Prime Time” in Biliary Tract Cancer. J. Hepatol. 2020, 73, 170–185. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Borad, M.J.; Kelley, R.K.; Wang, Y.; Abdel-Wahab, R.; Meric-Bernstam, F.; Baggerly, K.A.; Kaseb, A.O.; Al-Shamsi, H.O.; Ahn, D.H.; et al. Cholangiocarcinoma With FGFR Genetic Aberrations: A Unique Clinical Phenotype. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Rizzato, M.; Brignola, S.; Munari, G.; Gatti, M.; Dadduzio, V.; Borga, C.; Bergamo, F.; Pellino, A.; Angerilli, V.; Mescoli, C.; et al. Prognostic Impact of FGFR2/3 Alterations in Patients with Biliary Tract Cancers Receiving Systemic Chemotherapy: The BITCOIN Study. Eur. J. Cancer 2022, 166, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Mas, L.; Perrier, A.; Coulet, F.; Bachet, J.-B. Cholangiocarcinomes avancés et gènes de fusion. Bull. Du Cancer 2022, 109, 11S28–11S34. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://Www.Fda.Gov/Drugs/Development-Approval-Process-Drugs/Drug-Approvals-and-Databases (accessed on 28 August 2023).
- Vogel, A.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, S.; Borad, M.; Gallinson, D.; Murphy, A.; et al. O-2 Pemigatinib for Previously Treated Locally Advanced or Metastatic Cholangiocarcinoma: Final Results from FIGHT-202. Ann. Oncol. 2022, 33, S379. [Google Scholar] [CrossRef]
- Javle, M.; Roychowdhury, S.; Kelley, R.K.; Sadeghi, S.; Macarulla, T.; Weiss, K.H.; Waldschmidt, D.-T.; Goyal, L.; Borbath, I.; El-Khoueiry, A.; et al. Infigratinib (BGJ398) in Previously Treated Patients with Advanced or Metastatic Cholangiocarcinoma with FGFR2 Fusions or Rearrangements: Mature Results from a Multicentre, Open-Label, Single-Arm, Phase 2 Study. Lancet Gastroenterol. Hepatol. 2021, 6, 803–815. [Google Scholar] [CrossRef]
- Facchinetti, F.; Hollebecque, A.; Bahleda, R.; Loriot, Y.; Olaussen, K.A.; Massard, C.; Friboulet, L. Facts and New Hopes on Selective FGFR Inhibitors in Solid Tumors. Clin. Cancer Res. 2020, 26, 764–774. [Google Scholar] [CrossRef]
- Goyal, L.; Shi, L.; Liu, L.Y.; Fece de la Cruz, F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.W.S.; et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079. [Google Scholar] [CrossRef]
- Cleary, J.M.; Raghavan, S.; Wu, Q.; Li, Y.Y.; Spurr, L.F.; Gupta, H.V.; Rubinson, D.A.; Fetter, I.J.; Hornick, J.L.; Nowak, J.A.; et al. FGFR2 Extracellular Domain In-Frame Deletions Are Therapeutically Targetable Genomic Alterations That Function as Oncogenic Drivers in Cholangiocarcinoma. Cancer Discov. 2021, 11, 2488–2505. [Google Scholar] [CrossRef] [PubMed]
- Varghese, A.M.; Patel, J.; Janjigian, Y.Y.; Meng, F.; Selcuklu, S.D.; Iyer, G.; Houck-Loomis, B.; Harding, J.J.; O’Reilly, E.M.; Abou-Alfa, G.K.; et al. Noninvasive Detection of Polyclonal Acquired Resistance to FGFR Inhibition in Patients With Cholangiocarcinoma Harboring FGFR2 Alterations. JCO Precis. Oncol. 2021, 5, PO.20.00178. [Google Scholar] [CrossRef]
- Krook, M.A.; Bonneville, R.; Chen, H.-Z.; Reeser, J.W.; Wing, M.R.; Martin, D.M.; Smith, A.M.; Dao, T.; Samorodnitsky, E.; Paruchuri, A.; et al. Tumor Heterogeneity and Acquired Drug Resistance in FGFR2-Fusion-Positive Cholangiocarcinoma through Rapid Research Autopsy. Mol. Case Stud. 2019, 5, a004002. [Google Scholar] [CrossRef] [PubMed]
- Krook, M.A.; Lenyo, A.; Wilberding, M.; Barker, H.; Dantuono, M.; Bailey, K.M.; Chen, H.-Z.; Reeser, J.W.; Wing, M.R.; Miya, J.; et al. Efficacy of FGFR Inhibitors and Combination Therapies for Acquired Resistance in FGFR2-Fusion Cholangiocarcinoma. Mol. Cancer Ther. 2020, 19, 847–857. [Google Scholar] [CrossRef]
- Rengan, A.K.; Denlinger, C.S. Robust Response to Futibatinib in a Patient With Metastatic FGFR-Addicted Cholangiocarcinoma Previously Treated Using Pemigatinib. Cancer Netw. 2022, 20, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Lynch, P.T.; Bai, X.; Hsiehchen, D. Oncogenic RAS Drives Resistance to Pemigatinib in Cholangiocarcinoma Harboring a FGFR2 Delins Disrupting Ligand Binding. JCO Precis. Oncol. 2023, 7, e2200340. [Google Scholar] [CrossRef]
- Goyal, L.; Meric-Bernstam, F.; Hollebecque, A.; Valle, J.W.; Morizane, C.; Karasic, T.B.; Abrams, T.A.; Furuse, J.; Kelley, R.K.; Cassier, P.A.; et al. Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma. N. Engl. J. Med. 2023, 388, 228–239. [Google Scholar] [CrossRef]
- Borad, M.J.; Schram, A.M.; Kim, R.D.; Kamath, S.D.; Sahai, V.; Dotan, E.; Kelley, R.K.; Ponz-Sarvise, M.; Oh, D.-Y.; Yachnin, J.; et al. Updated Dose Escalation Results for ReFocus, a First-in-Human Study of Highly Selective FGFR2 Inhibitor RLY-4008 in Cholangiocarcinoma and Other Solid Tumors. J. Clin. Oncol. 2023, 41, 4009. [Google Scholar] [CrossRef]
- Park, J.O.; Meng, D.T.W.; Hollebecque, A.; Borad, M.; Goyal, L.; Schram, A.; Cassier, P.; Kamath, S.D.; Dotan, E.; Kim, R.; et al. 76MO Efficacy of RLY-4008, a Highly Selective FGFR2 Inhibitor in Patients (Pts) with an FGFR2-Fusion or Rearrangement (f/r), FGFR Inhibitor (FGFRi)-Naïve Cholangiocarcinoma (CCA): ReFocus Trial. Ann. Oncol. 2022, 33, S1461–S1462. [Google Scholar] [CrossRef]
- Guo, Y.; Yuan, C.; Ding, W.; Gao, Y.; Zhu, X.; Ying, J.; Sun, M.; Zhang, J.; Zhuang, Z.; Huang, Y.; et al. Gunagratinib, a Highly Selective Irreversible FGFR Inhibitor, in Patients with Previously Treated Locally Advanced or Metastatic Cholangiocarcinoma Harboring FGFR Pathway Alterations: A Phase IIa Dose-Expansion Study. J. Clin. Oncol. 2023, 41, 572. [Google Scholar] [CrossRef]
- Ouali, K.; Pellat, A.; Cohen, R.; Svrcek, M.; Penault-Llorca, F.; André, T. Fusions NTRK: Une Nouvelle Piste Dans Les Cancers Digestifs ? Bull. Du Cancer 2020, 107, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Boilève, A.; Verlingue, L.; Hollebecque, A.; Boige, V.; Ducreux, M.; Malka, D. Rare Cancer, Rare Alteration: The Case of NTRK Fusions in Biliary Tract Cancers. Expert Opin. Investig. Drugs 2021, 30, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK Gene Fusions as Novel Targets of Cancer Therapy across Multiple Tumour Types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in Patients with TRK Fusion-Positive Solid Tumours: A Pooled Analysis of Three Phase 1/2 Clinical Trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in Patients with Advanced or Metastatic NTRK Fusion-Positive Solid Tumours: Integrated Analysis of Three Phase 1-2 Trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Valery, M.; Facchinetti, F.; Malka, D.; Ducreux, M.; Friboulet, L.; Hollebecque, A. Cholangiocarcinoma with STRN-ALK Translocation Treated with ALK Inhibitors. Dig. Liver Dis. 2021, 53, 1664–1665. [Google Scholar] [CrossRef]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef]
- Zhu, A.X.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.T.; Borad, M.J.; Bridgewater, J.A.; et al. Final Overall Survival Efficacy Results of Ivosidenib for Patients With Advanced Cholangiocarcinoma With IDH1 Mutation: The Phase 3 Randomized Clinical ClarIDHy Trial. JAMA Oncol. 2021, 7, 1669–1677. [Google Scholar] [CrossRef]
- Subbiah, V.; Lassen, U.; Élez, E.; Italiano, A.; Curigliano, G.; Javle, M.; de Braud, F.; Prager, G.W.; Greil, R.; Stein, A.; et al. Dabrafenib plus Trametinib in Patients with BRAFV600E-Mutated Biliary Tract Cancer (ROAR): A Phase 2, Open-Label, Single-Arm, Multicentre Basket Trial. Lancet Oncol. 2020, 21, 1234–1243. [Google Scholar] [CrossRef]
- Maio, M.; Ascierto, P.A.; Manzyuk, L.; Motola-Kuba, D.; Penel, N.; Cassier, P.A.; Bariani, G.M.; De Jesus Acosta, A.; Doi, T.; Longo, F.; et al. Pembrolizumab in Microsatellite Instability High or Mismatch Repair Deficient Cancers: Updated Analysis from the Phase II KEYNOTE-158 Study. Ann. Oncol. 2022, 33, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Borad, M.J.; Azad, N.S.; Kurzrock, R.; Abou-Alfa, G.K.; George, B.; Hainsworth, J.; Meric-Bernstam, F.; Swanton, C.; Sweeney, C.J.; et al. Pertuzumab and Trastuzumab for HER2-Positive, Metastatic Biliary Tract Cancer (MyPathway): A Multicentre, Open-Label, Phase 2a, Multiple Basket Study. Lancet Oncol. 2021, 22, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Ohba, A.; Morizane, C.; Ueno, M.; Kobayashi, S.; Kawamoto, Y.; Komatsu, Y.; Ikeda, M.; Sasaki, M.; Okano, N.; Furuse, J.; et al. Multicenter Phase II Trial of Trastuzumab Deruxtecan for HER2-Positive Unresectable or Recurrent Biliary Tract Cancer: HERB Trial. Future Oncol. 2022, 18, 2351–2360. [Google Scholar] [CrossRef]
- Harding, J.J.; Fan, J.; Oh, D.-Y.; Choi, H.J.; Kim, J.W.; Chang, H.-M.; Bao, L.; Sun, H.-C.; Macarulla, T.; Xie, F.; et al. Zanidatamab for HER2-Amplified, Unresectable, Locally Advanced or Metastatic Biliary Tract Cancer (HERIZON-BTC-01): A Multicentre, Single-Arm, Phase 2b Study. Lancet Oncol. 2023, 24, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.S.; Spira, A.I.; Yaeger, R.; Buchschacher, G.L.; McRee, A.J.; Sabari, J.K.; Johnson, M.L.; Barve, M.A.; Hafez, N.; Velastegui, K.; et al. KRYSTAL-1: Updated Activity and Safety of Adagrasib (MRTX849) in Patients (Pts) with Unresectable or Metastatic Pancreatic Cancer (PDAC) and Other Gastrointestinal (GI) Tumors Harboring a KRASG12C Mutation. JCO 2022, 40, 519. [Google Scholar] [CrossRef]
- Koeberle, D.; Fritsch, R. Targeting HER2 in Biliary Tract Carcinomas: Challenges and Opportunities. Oncol. Res. Treat. 2021, 44, 1–3. [Google Scholar] [CrossRef]
- Nam, A.-R.; Kim, J.-W.; Cha, Y.; Ha, H.; Park, J.E.; Bang, J.-H.; Jin, M.H.; Lee, K.-H.; Kim, T.-Y.; Han, S.-W.; et al. Therapeutic Implication of HER2 in Advanced Biliary Tract Cancer. Oncotarget 2016, 7, 58007–58021. [Google Scholar] [CrossRef]
- Cortés, J.; Kim, S.-B.; Chung, W.-P.; Im, S.-A.; Park, Y.H.; Hegg, R.; Kim, M.H.; Tseng, L.-M.; Petry, V.; Chung, C.-F.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. N. Engl. J. Med. 2022, 386, 1143–1154. [Google Scholar] [CrossRef]
- Shitara, K.; Bang, Y.-J.; Iwasa, S.; Sugimoto, N.; Ryu, M.-H.; Sakai, D.; Chung, H.-C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N. Engl. J. Med. 2020, 382, 2419–2430. [Google Scholar] [CrossRef]
- Shitara, K.; Seraj, J.; Franke, F.A.; Kawaguchi, Y.; Shen, L.; Kamio, T.; Meinhardt, G.; Tabernero, J. 1436TiP Trastuzumab Deruxtecan (T-DXd) in Patients (Pts) with HER2-Positive Gastric Cancer (GC) or Gastroesophageal Junction (GEJ) Adenocarcinoma Who Have Progressed on or after a Trastuzumab-Containing Regimen (DESTINY-Gastric04, DG-04): A Randomized Phase III Study. Ann. Oncol. 2021, 32, S1073. [Google Scholar] [CrossRef]
- Salem, M.; El-Refai, S.; Sha, W.; Grothey, A.; Puccini, A.; George, T.; Hwang, J.; Kadakia, K.; Musselwhite, L.; Cutsem, E.V.; et al. O-3 Characterization of KRAS Mutation Variants and Prevalence of KRAS-G12C in Gastrointestinal Malignancies. Ann. Oncol. 2021, 32, S218. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.-H.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non–Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Weiss, J.; Pelster, M.S.; Spira, A.I.; Barve, M.; Ou, S.-H.I.; Leal, T.A.; Bekaii-Saab, T.S.; Paweletz, C.P.; Heavey, G.A.; et al. Adagrasib with or without Cetuximab in Colorectal Cancer with Mutated KRAS G12C. N. Engl. J. Med. 2023, 388, 44–54. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and Trametinib versus Dabrafenib and Placebo for Val600 BRAF-Mutant Melanoma: A Multicentre, Double-Blind, Phase 3 Randomised Controlled Trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Hashemi, S.M.S.; Mazieres, J.; Kim, T.M.; Quoix, E.; Souquet, P.-J.; Barlesi, F.; Baik, C.; et al. Phase 2 Study of Dabrafenib Plus Trametinib in Patients With BRAF V600E-Mutant Metastatic NSCLC: Updated 5-Year Survival Rates and Genomic Analysis. J. Thorac. Oncol. 2022, 17, 103–115. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- Rizzo, A.; Federico, A.D.; Ricci, A.D.; Frega, G.; Palloni, A.; Pagani, R.; Tavolari, S.; Marco, M.D.; Brandi, G. Targeting BRAF-Mutant Biliary Tract Cancer: Recent Advances and Future Challenges. Cancer Control 2020, 27. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Winkelmann, R.; Schneider, M.; Hartmann, S.; Schnitzbauer, A.A.; Zeuzem, S.; Peveling-Oberhag, J.; Hansmann, M.L.; Walter, D. Microsatellite Instability Occurs Rarely in Patients with Cholangiocarcinoma: A Retrospective Study from a German Tertiary Care Hospital. Int. J. Mol. Sci. 2018, 19, 1421. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch-Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Spizzo, G.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; Arora, S.P.; Khushman, M.; Salem, M.E.; Battaglin, F.; et al. Molecular Profile of BRCA-Mutated Biliary Tract Cancers. ESMO Open 2020, 5, e000682. [Google Scholar] [CrossRef]
- Ricci, A.D.; Rizzo, A.; Bonucci, C.; Tober, N.; Palloni, A.; Mollica, V.; Maggio, I.; Deserti, M.; Tavolari, S.; Brandi, G. PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block? Medicines 2020, 7, E54. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib Tablets as Maintenance Therapy in Patients with Platinum-Sensitive, Relapsed Ovarian Cancer and a BRCA1/2 Mutation (SOLO2/ENGOT-Ov21): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Tutt, A.N.J.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, J.; Qin, S.K.; Hua, H.Q. Treatment with Olaparib Monotherapy for BRCA2-Mutated Refractory Intrahepatic Cholangiocarcinoma: A Case Report. OncoTargets Ther. 2018, 11, 5957–5962. [Google Scholar] [CrossRef]
- Academic and Community Cancer Research United A Phase II Study of Olaparib in Patients With Advanced Biliary Tract Cancer With Aberrant DNA Repair Gene Mutations. 2022. Available online: https://clinicaltrials.gov (accessed on 28 August 2023).
- Nam, A.-R.; Yoon, J.; Jin, M.-H.; Bang, J.-H.; Oh, K.-S.; Seo, H.-R.; Kim, J.-M.; Kim, T.-Y.; Oh, D.-Y. ATR Inhibition Amplifies Antitumor Effects of Olaparib in Biliary Tract Cancer. Cancer Lett. 2021, 516, 38–47. [Google Scholar] [CrossRef]
- Mody, K.; Jain, P.; El-Refai, S.M.; Azad, N.S.; Zabransky, D.J.; Baretti, M.; Shroff, R.T.; Kelley, R.K.; El-Khouiery, A.B.; Hockenberry, A.J.; et al. Clinical, Genomic, and Transcriptomic Data Profiling of Biliary Tract Cancer Reveals Subtype-Specific Immune Signatures. JCO Precis. Oncol. 2022, 6, e2100510. [Google Scholar] [CrossRef]


{kind=link}
| Somatic Alteration | Primitive Tumour Location and Frequency | Study | Drug (s) | Results |
|---|---|---|---|---|
| FGFR fusion | 15% of iCCA | FIGHT-202, [40] (phase II), ≥L2 | Pemigatinib (FGFR 1–3 inhibitor) | ORR 37% DCR 82% mPFS 7 mo, mOS 17.5 mo |
| FOENIX-CCA2, [50] (phase II), ≥L2 | Futibatinib (Pan-FGFR inhibitor) | ORR 42%, DCR 83% mPFS 8.9 mo, mOS 20 mo | ||
| ReFocus, [52] (phase I/II), ≥L2 | RLY-4008 (Pan- FGFR inhibitor) | ORR 53% DCR 94% mPFS 6.9 mo | ||
| IDH1 mutation | 15% of iCCA | ClarIDHy, [14] (phase III) L2–L3 | Ivosidenib | ORR 2%; DCR 51% mPFS 2.7 mo, mOS 10.8 mo |
| BRAF mutation | 5% of iCCA | ROAR, [63] (phase II) ≥L2 | Dabrafenib + Trametinib | ORR 42% |
| MSI | 5% of BTC | Keynote-158, [64] (phase II multi cohorte) ≥L2 | Pembrolizumab | ORR 40.9% |
| BRCA1/2 mutation | 3–5% of BTC | NCT 04042831 (phase II, pending) ≥L2 | Olaparib | Pending |
| HER2 (mutation and amplification) | 5% of eCCA 10–15% of GBC | My pathway, [65] (phase II basket) ≥L2 | Trastuzumab + pertuzumab | 11 pts with BTC, 8 amplifications, 3 mutations: ORR 3/8 and 1/3; mPFS 4.2 mo and 2.8 mo, respectively |
| JMA-IIA00423, [66] (phase II) ≥L2 | Trastuzumab-deruxtecan | ORR 36.4%; mPFS 5.1 mo, mOS 7.1 mo | ||
| HERIZON-BTC 01, [67] (phase IIb) | Zanidatamab | ORR 41.3% mPFS 5.5 mo | ||
| KRAS G12C mutation | 2% of KRAS mutations KRAS mutations: 20% of iCCA, 40–50% of eCCA | Krystal-I, [68] (phase II multi cohorte) ≥L2 | Adagrasib | ORR 50%, mPFS 7.9 mo |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valery, M.; Vasseur, D.; Fachinetti, F.; Boilève, A.; Smolenschi, C.; Tarabay, A.; Antoun, L.; Perret, A.; Fuerea, A.; Pudlarz, T.; et al. Targetable Molecular Alterations in the Treatment of Biliary Tract Cancers: An Overview of the Available Treatments. Cancers 2023, 15, 4446. https://doi.org/10.3390/cancers15184446
Valery M, Vasseur D, Fachinetti F, Boilève A, Smolenschi C, Tarabay A, Antoun L, Perret A, Fuerea A, Pudlarz T, et al. Targetable Molecular Alterations in the Treatment of Biliary Tract Cancers: An Overview of the Available Treatments. Cancers. 2023; 15(18):4446. https://doi.org/10.3390/cancers15184446
Chicago/Turabian StyleValery, Marine, Damien Vasseur, Francesco Fachinetti, Alice Boilève, Cristina Smolenschi, Anthony Tarabay, Leony Antoun, Audrey Perret, Alina Fuerea, Thomas Pudlarz, and et al. 2023. "Targetable Molecular Alterations in the Treatment of Biliary Tract Cancers: An Overview of the Available Treatments" Cancers 15, no. 18: 4446. https://doi.org/10.3390/cancers15184446
APA StyleValery, M., Vasseur, D., Fachinetti, F., Boilève, A., Smolenschi, C., Tarabay, A., Antoun, L., Perret, A., Fuerea, A., Pudlarz, T., Boige, V., Hollebecque, A., & Ducreux, M. (2023). Targetable Molecular Alterations in the Treatment of Biliary Tract Cancers: An Overview of the Available Treatments. Cancers, 15(18), 4446. https://doi.org/10.3390/cancers15184446