

Targeting FGFRs Using PD173074 as a Novel Therapeutic Strategy in Cholangiocarcinoma

 ,
,  ,
,  , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Pre-Processing
2.2. Enrichment Analysis
2.3. Survival Analysis
2.4. Immunohistochemistry
2.5. Cell Line Culture
2.6. Cell Viability Assays
2.7. Colony Formation Assays
2.8. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.9. Western Blotting Analysis
2.10. Human Phospho-Protein Proteome Profiler Array
2.11. Caspase 3 Activity Assay
2.12. Flow Cytometry Apoptosis Assay
2.13. Synergy
2.14. 3-D Tumour Growth Assay
3. Results
3.1. Aberrant Expression of FGFRs Was Observed in CCA Tissues Using an Integrated Bioinformatics Approach
3.2. High Expression of FGFRs Is Associated with Cancer Hallmark Pathways in CCA
3.3. Clinical Relevance of FGFR Gene Signature Suggests That FGFR Expression Can Be Used to Stratify CCA Patients According to Risk
3.4. PD173074 Is a Potential Candidate for FGFR Inhibition in CCA
3.5. Sensitivity to PD173074 in CCA Cell Lines
3.6. PD173074 Induces Apoptosis in CCA
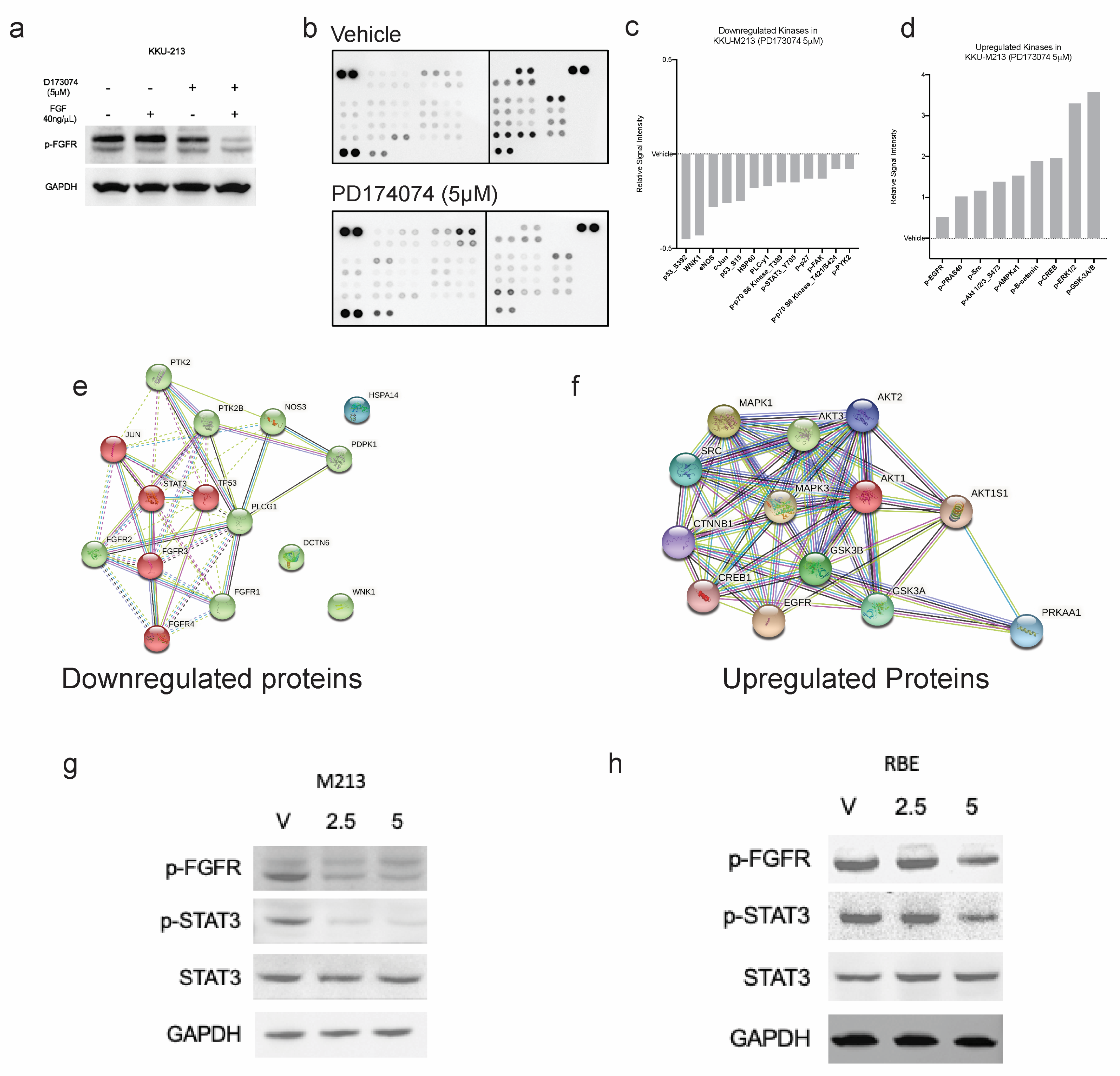
3.7. PD173074 Blocks FGF-Stimulated FGFRs and Further Downstream Signalling
3.8. Combination Treatment with Erlotinib Increases the Sensitivity of CCA Cells to Inhibition by PD173074
3.9. Combination Treatment in a 3-D Setting with the Presence of Tumour Stromal Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- DeOliveira, M.L.; Cunningham, S.C.; Cameron, J.L.; Kamangar, F.; Winter, J.M.; Lillemoe, K.D.; Choti, M.A.; Yeo, C.J.; Schulick, R.D. Cholangiocarcinoma: Thirty-one-year experience with 564 patients at a single institution. Ann. Surg. 2007, 245, 755–762. [Google Scholar] [CrossRef]
- Tawarungruang, C.; Khuntikeo, N.; Chamadol, N.; Laopaiboon, V.; Thuanman, J.; Thinkhamrop, K.; Kelly, M.; Thinkhamrop, B. Survival after surgery among patients with cholangiocarcinoma in Northeast Thailand according to anatomical and morphological classification. BMC Cancer 2021, 21, 497. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.M.; Vogel, A.; Arrese, M.; Balderramo, D.C.; Valle, J.W.; Banales, J.M. Next-Generation Biomarkers for Cholangiocarcinoma. Cancers 2021, 13, 3222. [Google Scholar] [CrossRef] [PubMed]
- Dabney, R.S.; Khalife, M.; Shahid, K.; Phan, A.T. Molecular pathways and targeted therapy in cholangiocarcinoma. Clin. Adv. Hematol. Oncol. 2019, 17, 630–637. [Google Scholar] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Borad, M.J.; Champion, M.D.; Egan, J.B.; Liang, W.S.; Fonseca, R.; Bryce, A.H.; McCullough, A.E.; Barrett, M.T.; Hunt, K.; Patel, M.D.; et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet. 2014, 10, e1004135. [Google Scholar] [CrossRef]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Yue, S.; Li, Y.; Chen, X.; Wang, J.; Li, M.; Chen, Y.; Wu, D. FGFR-TKI resistance in cancer: Current status and perspectives. J. Hematol. Oncol. 2021, 14, 23. [Google Scholar] [CrossRef]
- Balasubramanian, B.; Venkatraman, S.; Janvilisri, T.; Suthiphongchai, T.; Jitkaew, S.; Sripa, J.; Tohtong, R. RTK25: A Comprehensive Molecular Profiling Strategy in Cholangiocarcinoma Using an Integrated Bioinformatics Approach. Pharmaceuticals 2021, 14, 898. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Crowe, A.R.; Yue, W. Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio Protoc. 2019, 9, e3465. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Dikic, I.; Sorokin, A.; Burgess, W.H.; Jaye, M.; Schlessinger, J. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol. Cell. Biol. 1996, 16, 977–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, J.J.G.; Lozano, E.; Herraez, E.; Asensio, M.; Di Giacomo, S.; Romero, M.R.; Briz, O.; Serrano, M.A.; Efferth, T.; Macias, R.I.R. Chemoresistance and chemosensitization in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- (FDA), T.F.a.D.A. FDA Approves Ivosidenib for Advanced or Metastatic Cholangiocarcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ivosidenib-advanced-or-metastatic-cholangiocarcinoma (accessed on 24 January 2022).
- (FDA), T.F.a.D.A. FDA Grants Accelerated Approval to Pemigatinib for Cholangiocarcinoma with an FGFR2 Rearrangement or Fusion. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pemigatinib-cholangiocarcinoma-fgfr2-rearrangement-or-fusion (accessed on 2 July 2022).
- Jain, A.; Borad, M.J.; Kelley, R.K.; Wang, Y.; Abdel-Wahab, R.; Meric-Bernstam, F.; Baggerly, K.A.; Kaseb, A.O.; Al-Shamsi, H.O.; Ahn, D.H.; et al. Cholangiocarcinoma With FGFR Genetic Aberrations: A Unique Clinical Phenotype. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.P.; Barr Fritcher, E.G.; Pestova, E.; Schulz, J.; Sitailo, L.A.; Vasmatzis, G.; Murphy, S.J.; McWilliams, R.R.; Hart, S.N.; Halling, K.C.; et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum. Pathol. 2014, 45, 1630–1638. [Google Scholar] [CrossRef]
- Al Zaid Siddiquee, K.; Turkson, J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell. Res. 2008, 18, 254–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatraman, S.; Balasubramanian, B.; Pongchaikul, P.; Tohtong, R.; Chutipongtanate, S. Molecularly Guided Drug Repurposing for Cholangiocarcinoma: An Integrative Bioinformatic Approach. Genes. 2022, 13, 271. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Abreu, M.T.; Pearson, A.; Campbell, J.; Shnyder, S.D.; Knowles, M.A.; Ashworth, A.; Turner, N.C. Parallel RNA interference screens identify EGFR activation as an escape mechanism in FGFR3-mutant cancer. Cancer Discov. 2013, 3, 1058–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Wang, H.; Xie, E.; Tang, B.; Mu, Q.; Song, Z.; Chen, J.; Wang, F.; Min, J. Rewiring ERBB3 and ERK signaling confers resistance to FGFR1 inhibition in gastrointestinal cancer harbored an ERBB3-E928G mutation. Protein Cell. 2020, 11, 915–920. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pFGFR | ||||
|---|---|---|---|---|
| Parameters | n | Low (n = 25) | High (n = 15) | p-Value |
| Age | ||||
| <58 | 7 | 3 | 4 | 0.4705 |
| >58 | 17 | 12 | 5 | 0.3730 |
| Gender | ||||
| Male | 18 | 10 | 8 | 0.2749 |
| Female | 6 | 5 | 1 | 0.3508 |
| Survival | ||||
| <1 year | 18 | 11 | 7 | 0.5023 |
| >1 year | 6 | 4 | 2 | 0.0177 * |
| IC50 (uM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Erlotinib | PD173074 | Erlotinib + PD173074 | |||||||
| - | +MSCs | +CAFs | - | +MSCs | +CAFs | - | +MSCs | +CAFs | |
| CCA5 | 6.759 | 3.639 | 6.134 | 5.476 | 6.927 | 9.237 | 7.397 | 2.448 | 2.287 |
| CCA6 | 10.38 | 18.41 | 10.51 | 100 | 17.16 | 100 | 1.668 | 8.179 | 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balasubramanian, B.; Yacqub-Usman, K.; Venkatraman, S.; Myint, K.Z.; Juengsamarn, J.; Sarkhampee, P.; Lertsawatvicha, N.; Sripa, J.; Kuakpaetoon, T.; Suriyonplengsaeng, C.; et al. Targeting FGFRs Using PD173074 as a Novel Therapeutic Strategy in Cholangiocarcinoma. Cancers 2023, 15, 2528. https://doi.org/10.3390/cancers15092528
Balasubramanian B, Yacqub-Usman K, Venkatraman S, Myint KZ, Juengsamarn J, Sarkhampee P, Lertsawatvicha N, Sripa J, Kuakpaetoon T, Suriyonplengsaeng C, et al. Targeting FGFRs Using PD173074 as a Novel Therapeutic Strategy in Cholangiocarcinoma. Cancers. 2023; 15(9):2528. https://doi.org/10.3390/cancers15092528
Chicago/Turabian StyleBalasubramanian, Brinda, Kiren Yacqub-Usman, Simran Venkatraman, Kyaw Zwar Myint, Jitlada Juengsamarn, Poowanai Sarkhampee, Nithi Lertsawatvicha, Jittiyawadee Sripa, Thiti Kuakpaetoon, Chinnawut Suriyonplengsaeng, and et al. 2023. "Targeting FGFRs Using PD173074 as a Novel Therapeutic Strategy in Cholangiocarcinoma" Cancers 15, no. 9: 2528. https://doi.org/10.3390/cancers15092528
APA StyleBalasubramanian, B., Yacqub-Usman, K., Venkatraman, S., Myint, K. Z., Juengsamarn, J., Sarkhampee, P., Lertsawatvicha, N., Sripa, J., Kuakpaetoon, T., Suriyonplengsaeng, C., Wongprasert, K., Grabowska, A. M., Bates, D. O., Janvilisri, T., & Tohtong, R. (2023). Targeting FGFRs Using PD173074 as a Novel Therapeutic Strategy in Cholangiocarcinoma. Cancers, 15(9), 2528. https://doi.org/10.3390/cancers15092528