Hydrodeoxygenation of Levulinic Acid Dimers on a Zirconia-Supported Ruthenium Catalyst

 , and
, and
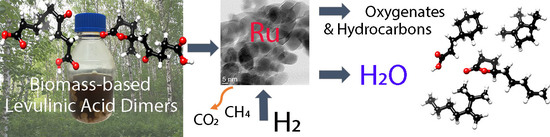
Abstract
:
1. Introduction
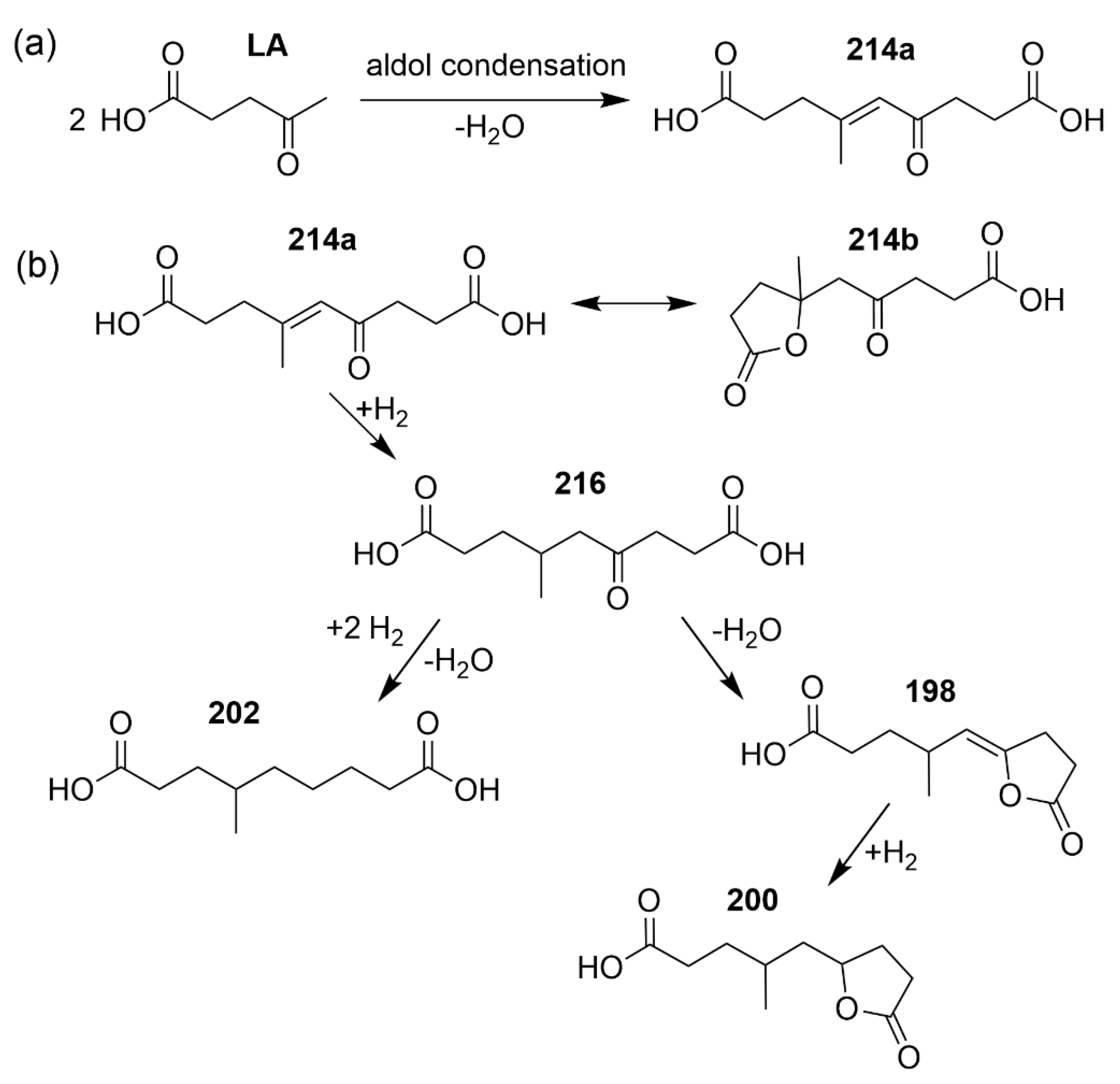
2. Results and Discussion
2.1. Catalyst Characterization
2.2. Relative Change of LA Dimers
2.3. Elemental Composition
2.4. Molecular Sizes of the Products
2.5. Product Composition by Infrared Spectroscopy
2.6. Product Composition by Nuclear Magnetic Resonance Spectroscopy
2.7. Light and Volatile Products
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation
3.3. Catalyst Characterization
3.4. HDO Experiments
3.5. Product Analytics
4. Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oh, Y.K.; Hwang, K.R.; Kim, C.; Kim, J.R.; Lee, J.S. Recent developments and key barriers to advanced biofuels: A short review. Bioresour. Technol. 2018, 257, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.N.; Goud, V.V.; Rout, P.K.; Dalai, A.K. Production of first and second generation biofuels: A comprehensive review. Renew. Sustain. Energy Rev. 2010, 14, 578–597. [Google Scholar] [CrossRef]
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass Volume I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas; National Renewable Energy Lab.: Golden, CO, USA, 2004; p. 76. [Google Scholar]
- Isikgor, F.H.; Becer, C.R. Lignocellulosic biomass: A sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef] [Green Version]
- Mascal, M.; Dutta, S.; Gandarias, I. Hydrodeoxygenation of the angelica lactone dimer, a cellulose-based feedstock: Simple, high-yield synthesis of branched C7-C10 gasoline-like hydrocarbons. Angew. Chem. Int. Ed. 2014, 53, 1854–1857. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.Q.; Upadhye, A.A.; Olcay, H.; Tompsett, G.A.; Jae, J.; Xing, R.; Alonso, D.M.; Wang, D.; Zhang, T.; Kumar, R.; et al. Production of renewable jet fuel range alkanes and commodity chemicals from integrated catalytic processing of biomass. Energy Environ. Sci. 2014, 7, 1500–1523. [Google Scholar] [CrossRef]
- Käldström, M.; Lindblad, M.; Lamminpää, K.; Wallenius, S.; Toppinen, S. Carbon Chain Length Increase Reactions of Platform Molecules Derived from C5 and C6 Sugars. Ind. Eng. Chem. Res. 2017, 56, 13356–13366. [Google Scholar] [CrossRef]
- Huber, G.W.; Chheda, J.N.; Barrett, C.J.; Dumesic, J.A. Production of Liquid Alkanes by Aqueous-Phase Processing of Biomass-Derived Carbohydrates. Science 2005, 308, 1446–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, R.M.; Liu, Z.Y.; Peter, M.; Dumesic, J.A. Liquid alkanes with targeted molecular weights from biomass-derived carbohydrates. ChemSusChem 2008, 1, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.; Chen, E.Y.X. Organocatalytic Cross-Coupling of Biofuranics to Multifunctional Difuranic C11 Building Blocks. ACS Sustain. Chem. Eng. 2016, 4, 4927–4936. [Google Scholar] [CrossRef]
- Zang, H.; Chen, E.Y.X. Organocatalytic upgrading of furfural and 5-hydroxymethyl furfural to C10 and C12 furoins with quantitative yield and atom-efficiency. Int. J. Mol. Sci. 2015, 16, 7143–7158. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Li, N.; Wang, Z.; Li, C.; Wang, A.; Wang, X.; Cong, Y.; Zhang, T. Synthesis of high-quality diesel with furfural and 2-methylfuran from hemicellulose. ChemSusChem 2012, 5, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; De La Torre, O.; Renz, M. Production of high quality diesel from cellulose and hemicellulose by the Sylvan process: Catalysts and process variables. Energy Environ. Sci. 2012, 5, 6328–6344. [Google Scholar] [CrossRef]
- Serrano-Ruiz, J.C.; Wang, D.; Dumesic, J.A. Catalytic upgrading of levulinic acid to 5-nonanone. Green Chem. 2010, 12, 574. [Google Scholar] [CrossRef]
- Serrano-Ruiz, J.C.; Braden, D.J.; West, R.M.; Dumesic, J.A. Conversion of cellulose to hydrocarbon fuels by progressive removal of oxygen. Appl. Catal. B Environ. 2010, 100, 184–189. [Google Scholar] [CrossRef]
- Bond, J.Q.; Alonso, D.M.; Wang, D.; West, R.M.; Dumesic, J.A. Integrated Catalytic Conversion of γ-Valerolactone to Liquid Alkenes for Transportation Fuels. Science 2010, 1110–1114. [Google Scholar] [CrossRef]
- Grilc, M.; Likozar, B. Levulinic acid hydrodeoxygenation, decarboxylation and oligmerization over NiMo/Al2O3 catalyst to bio-based value-added chemicals: Modelling of mass transfer, thermodynamics and micro-kinetics. Chem. Eng. J. 2017, 330, 383–397. [Google Scholar] [CrossRef]
- Blessing, R.W.; Petrus, L. A Catalytic Process for the Dimerization of Levulinic Acid and the Preparation of Diesters from these Dimers Obtainable by such Process. WO 2006/056591 A1, 1 June 2006. [Google Scholar]
- Faba, L.; Díaz, E.; Ordóñez, S. Base-Catalyzed Condensation of Levulinic Acid: A New Biorefinery Upgrading Approach. ChemCatChem 2016, 8, 1490–1494. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.; Nielsen, M.M.; Wang, H.; Chen, C.; Xu, J.; Wang, Y.; Deng, T.; Hou, X. Efficient C-C Bond Formation between Two Levulinic Acid Molecules to Produce C 10 Compounds with the Cooperation Effect of Lewis and Brønsted Acids. ACS Sustain. Chem. Eng. 2018, 6, 5708–5711. [Google Scholar] [CrossRef]
- Amarasekara, A.S.; Wiredu, B.; Grady, T.L.; Obregon, R.G.; Margetić, D. Solid acid catalyzed aldol dimerization of levulinic acid for the preparation of C10 renewable fuel and chemical feedstocks. Catal. Commun. 2019, 124, 6–11. [Google Scholar] [CrossRef]
- Cornils, B.; Lappe, P. Dicarboxylic Acids, Aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; pp. 1–18. [Google Scholar]
- Van den Brink, P.J.; von Hebel, K.L.; Lange, J.-P.; Petrus, L. A Process for the Hydrogenation of a Lactone or of a Carboxylic Acid or an Ester having a Gamma-Carbonyl Group. WO2006067171, 29 June 2006. [Google Scholar]
- González Escobedo, J.L.; Mäkelä, E.; Braunschweiler, A.; Lehtonen, J.; Lindblad, M.; Puurunen, R.L.; Karinen, R. Solvent-free Hydrodeoxygenation of γ-Nonalactoneon on Noble Metal Catalysts Supported on Zirconia. Top. Catal. 2018, 62, 724–737. [Google Scholar] [CrossRef] [Green Version]
- Chia, M.; Pagán-Torres, Y.J.; Hibbitts, D.; Tan, Q.; Pham, H.N.; Datye, A.K.; Neurock, M.; Davis, R.J.; Dumesic, J.A. Selective hydrogenolysis of polyols and cyclic ethers over bifunctional surface sites on rhodium-rhenium catalysts. J. Am. Chem. Soc. 2011, 133, 12675–12689. [Google Scholar] [CrossRef] [PubMed]
- Bie, Y.; Gutierrez, A.; Viljava, T.R.; Kanervo, J.M.; Lehtonen, J. Hydrodeoxygenation of Methyl Heptanoate over Noble Metal Catalysts: Catalyst Screening and Reaction Network. Ind. Eng. Chem. Res. 2013, 52, 11544–11551. [Google Scholar] [CrossRef]
- Jung, K.T.; Bell, A.T. The effects of synthesis and pretreatment conditions on the bulk structure and surface properties of zirconia. J. Mol. Catal. A Chem. 2000, 163, 27–42. [Google Scholar] [CrossRef]
- van Krevelen, D.W. Graphical-statistical method for the study of structure and reaction processes of coal. Fuel 1950, 29, 269–283. [Google Scholar]
- Leitner, W.; Klankermayer, J.; Pischinger, S.; Pitsch, H.; Kohse-Höinghaus, K. Advanced Biofuels and Beyond: Chemistry Solutions for Propulsion and Production. Angew. Chem. Int. Ed. 2017, 56, 5412–5452. [Google Scholar] [CrossRef] [PubMed]
- Field, L.D.; Sternhell, S.; Kalman, J.R. Organic Structures from Spectra, 5th ed.; Field, L.D., Sternhell, S., Kalman, J.R., Eds.; John Wiley & Sons, Ltd.: West Sussex, UK, 2013; p. 497. [Google Scholar]
- Beauchamp, P. Spectroscopy Tables. Available online: https://www.cpp.edu/~psbeauchamp/pdf/spec_ir_nmr_spectra_tables.pdf (accessed on 2 December 2019).
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. In Encyclopedia of Analytic Chemistry: Applications, Theory and Instrumentation; Meyers, R.A., Ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2006; pp. 10815–10837. [Google Scholar]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Balci, M. Basic 1H-and 13C-NMR Spectroscopy; Elsevier B.V.: Amsterdam, The Netherlands, 2005; p. 419. [Google Scholar]
- Myllyoja, J.; Lindblad, M.; Käldström, M.; Piilola, R.; Ikonen, E. Renewable Hydrocarbons, Method for Producing the Same and the Use Thereof. EP3187482A1, 29 December 2015. [Google Scholar]
- Minachev, K.M.; Shuikin, N.I.; Rozhdestvenskaya, I.D. Hydrogenation and dehydrogenation of hydrocarbons in presence of ruthenium and rhodium catalysts of low metal content. Bull. Acad. Sci. Ussr Div. Chem. Sci. 1954, 3, 277–281. [Google Scholar] [CrossRef]
- Pirogova, G.N.; Rimar’, N.N.; Kalinina, G.E. Synergistic effect for bimetallic Ru-Tc catalysts of cyclohexane dehydrogenation. Russ. Chem. Bull. 1996, 45, 1857–1861. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Shen, X.; Garces, L.; Ding, Y.; Laubernds, K.; Zerger, R.P.; Aindow, M.; Neth, E.J.; Suib, S.L. Behavior of H2 chemisorption on Ru / TiO2 surface and its application in evaluation of Ru particle sizes compared with TEM and XRD analyses. Appl. Catal. A Gen. 2008, 335, 187–195. [Google Scholar] [CrossRef]
- Bergeret, G.; Gallezot, P. Particle Size and Dispersion Measurements. In Handbook of Heteregeneous Catalysis; Ertl, G., Knözinger, H., Schuth, F., Weitkamp, J., Eds.; WILEY-VHC Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 738–746. ISBN 978-3-527-31241-2. [Google Scholar]
- Guerrero, H.; Lafuente, C.; Royo, F.; Lomba, L.; Giner, B. PρT behavior of several chemicals from biomass. Energy Fuels 2011, 25, 3009–3013. [Google Scholar] [CrossRef]
- Scanlon, J.T.; Willis, D.E. Calculation of Flame Ionization Detector Relative Response Factors Using the Effective Carbon Number Concept. J. Chromatogr. Sci. 1985, 23, 333–340. [Google Scholar] [CrossRef]
- Fogler, H.S. Elements of Chemical Reaction Engineering, 4th ed.; Pearson Education, Inc.: Westford, MA, USA, 2006; pp. 66–67. ISBN 0-13-127839-8. [Google Scholar]
- Kapteijn, F.; Gascon, J.; Nijhuis, T.A. Catalytic Reaction Engineering. In Catalysis An Integrated Textbook for Students; Hanefeld, U., Lefferts, L., Eds.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2008; pp. 221–271. ISBN 978-3-527-34159-7. [Google Scholar]
- Boudart, M. Turnover Rates in Heterogeneous Catalysis. Chem. Rev. 1995, 95, 661–666. [Google Scholar] [CrossRef]
- Deutschmann, O.; Knözinger, H.; Kochloefl, K.; Turek, T. Heterogeneous Catalysis and Solid Catalysts, 1. Fundamentals. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 565–582. ISBN 9783527306732. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | XRF | Physisorption | Chemisorption | ||||
|---|---|---|---|---|---|---|---|
| Metal content | SBET 1 (m2 g−1) | Pore volume 2 (cm3 g−1) | Average pore diameter 2 (nm) | Irreversible adsorption capacity (µmolgas gcat−1) | Dispersion | Average particle size (nm) | |
| ZrO2 | - | 67 | 0.18 | 13 | - | - | - |
| Ru/ZrO2 | 3.6% | 70 | 0.21 | 11 | 43.9 | 25 | 3.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mäkelä, E.; González Escobedo, J.L.; Lindblad, M.; Käldström, M.; Meriö-Talvio, H.; Jiang, H.; Puurunen, R.L.; Karinen, R. Hydrodeoxygenation of Levulinic Acid Dimers on a Zirconia-Supported Ruthenium Catalyst. Catalysts 2020, 10, 200. https://doi.org/10.3390/catal10020200
Mäkelä E, González Escobedo JL, Lindblad M, Käldström M, Meriö-Talvio H, Jiang H, Puurunen RL, Karinen R. Hydrodeoxygenation of Levulinic Acid Dimers on a Zirconia-Supported Ruthenium Catalyst. Catalysts. 2020; 10(2):200. https://doi.org/10.3390/catal10020200
Chicago/Turabian StyleMäkelä, Eveliina, José Luis González Escobedo, Marina Lindblad, Mats Käldström, Heidi Meriö-Talvio, Hua Jiang, Riikka L. Puurunen, and Reetta Karinen. 2020. "Hydrodeoxygenation of Levulinic Acid Dimers on a Zirconia-Supported Ruthenium Catalyst" Catalysts 10, no. 2: 200. https://doi.org/10.3390/catal10020200
APA StyleMäkelä, E., González Escobedo, J. L., Lindblad, M., Käldström, M., Meriö-Talvio, H., Jiang, H., Puurunen, R. L., & Karinen, R. (2020). Hydrodeoxygenation of Levulinic Acid Dimers on a Zirconia-Supported Ruthenium Catalyst. Catalysts, 10(2), 200. https://doi.org/10.3390/catal10020200