An Eco-Friendly Fluidizable FexOy/CaO-γ-Al2O3 Catalyst for Tar Cracking during Biomass Gasification




Abstract
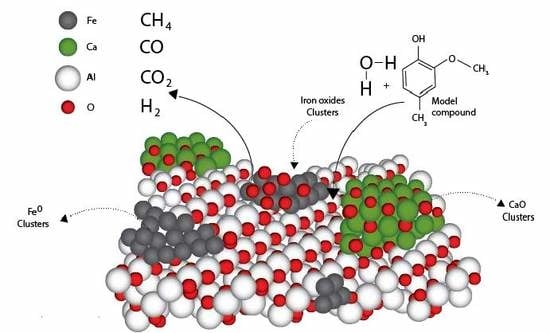
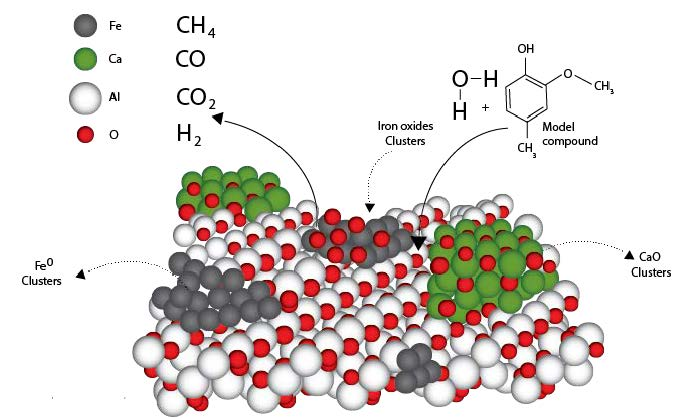
:
1. Introduction
2. Results and Discussion
2.1. Constraints in Catalyst Design
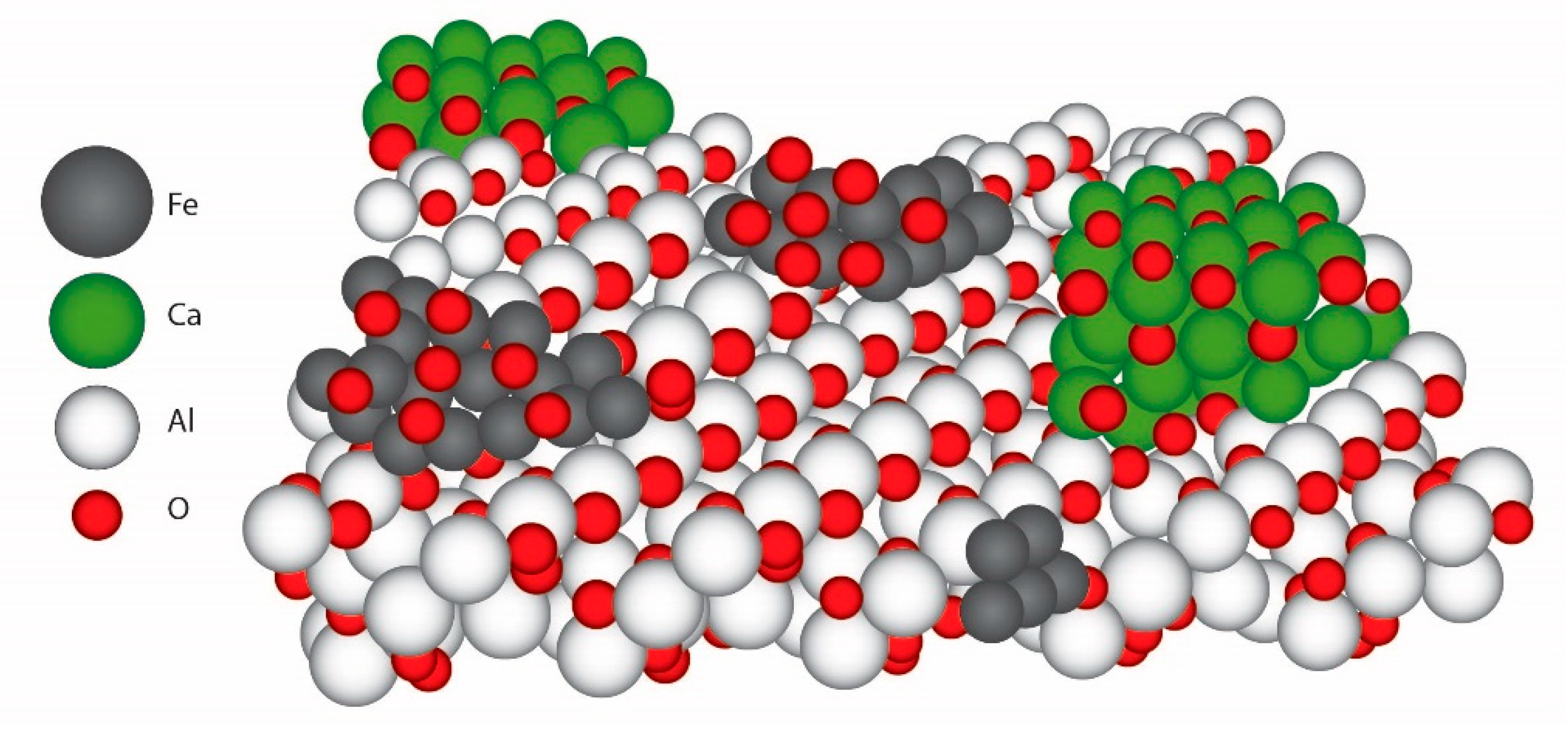
2.2. Design of the Proposed Catalyst
2.3. Characterization of the Prepared Catalyst
2.3.1. Catalyst’s Structural Properties
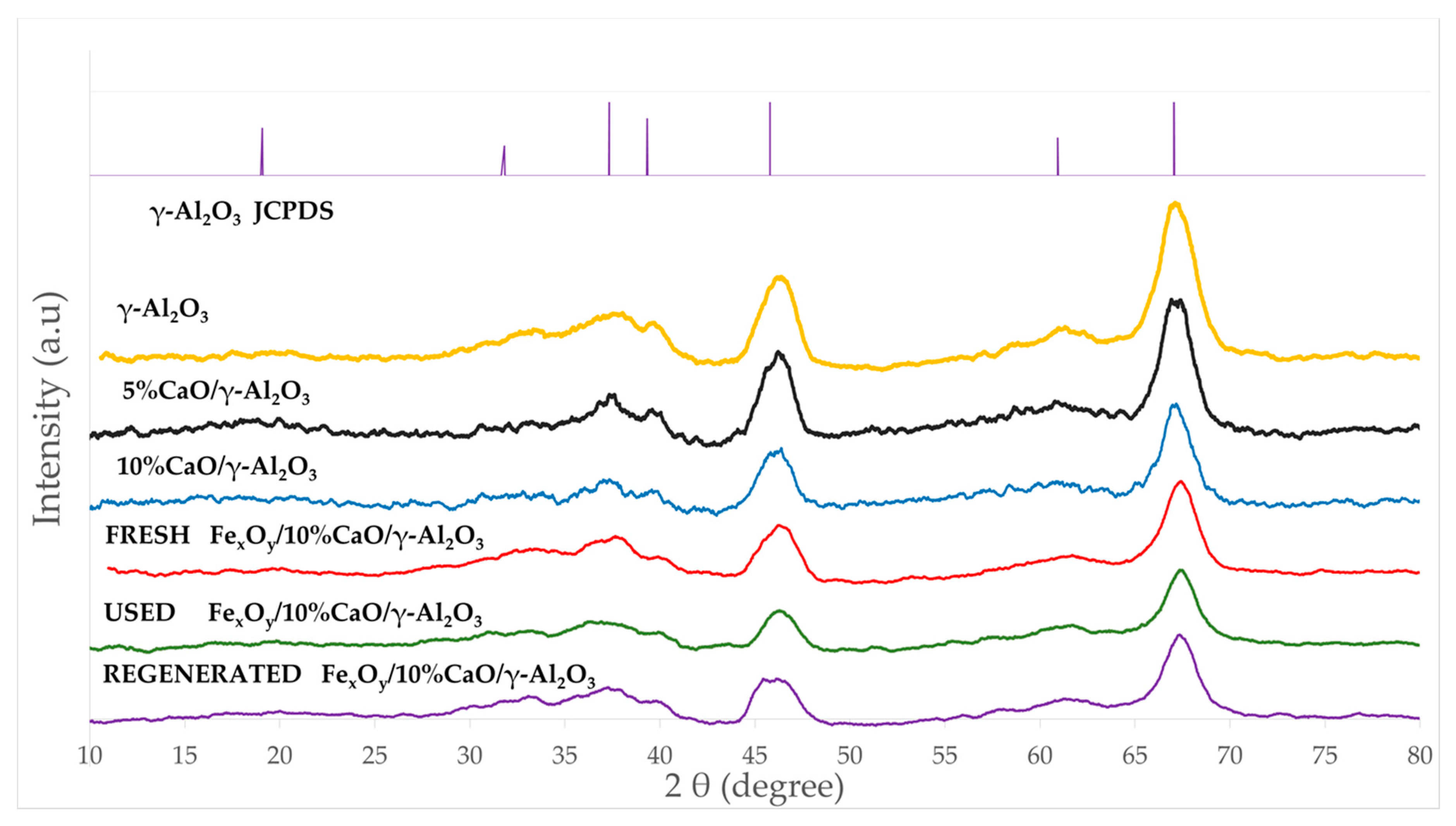
2.3.2. Phases Present in the Iron Oxide/CaO/γ-Al2O3 Catalyst
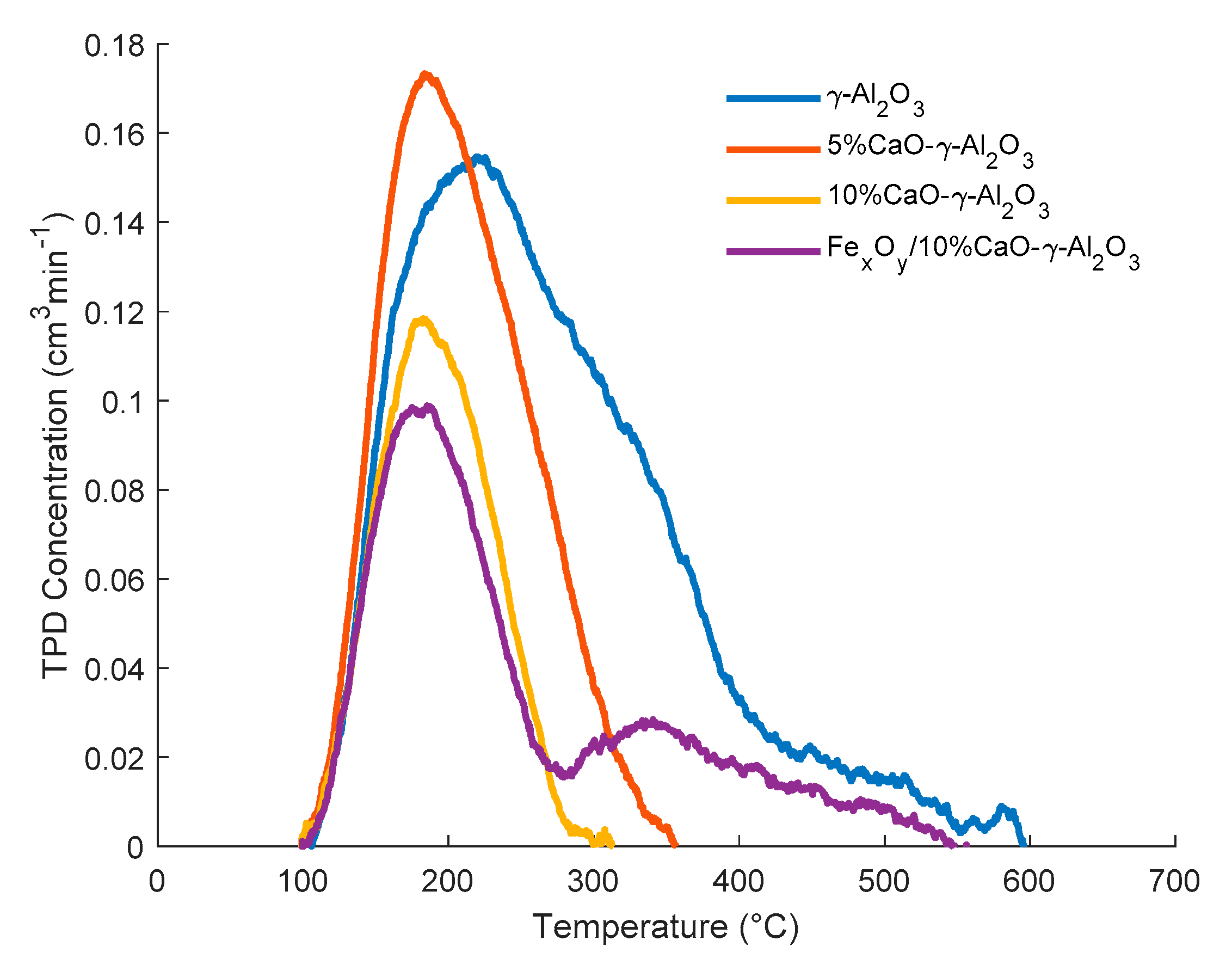
2.3.3. Influence of CaO and Iron Dopants on Acidity
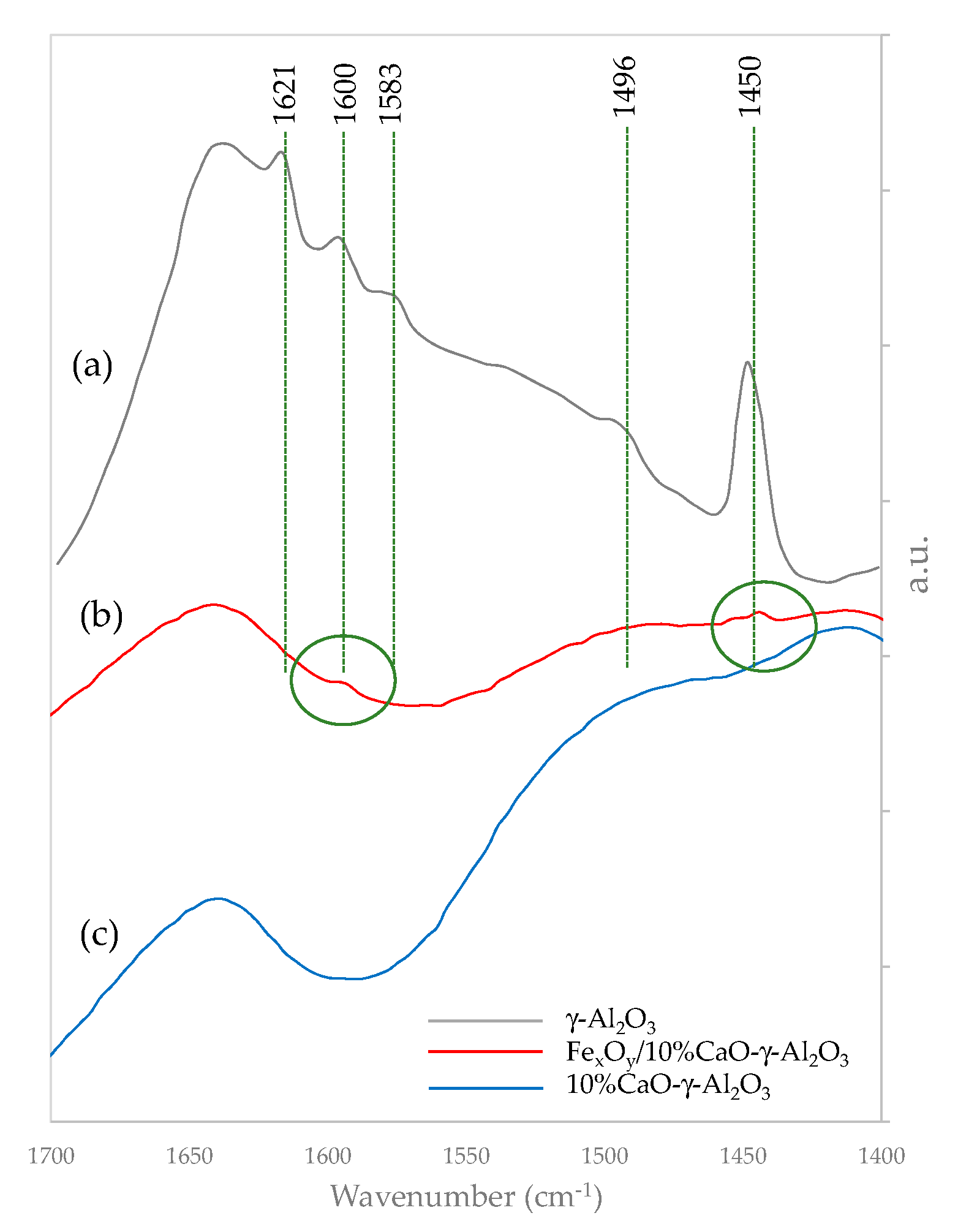
2.3.4. Pyridine Desorption Infrared Spectroscopy
2.3.5. Chemical State of Catalyst Species
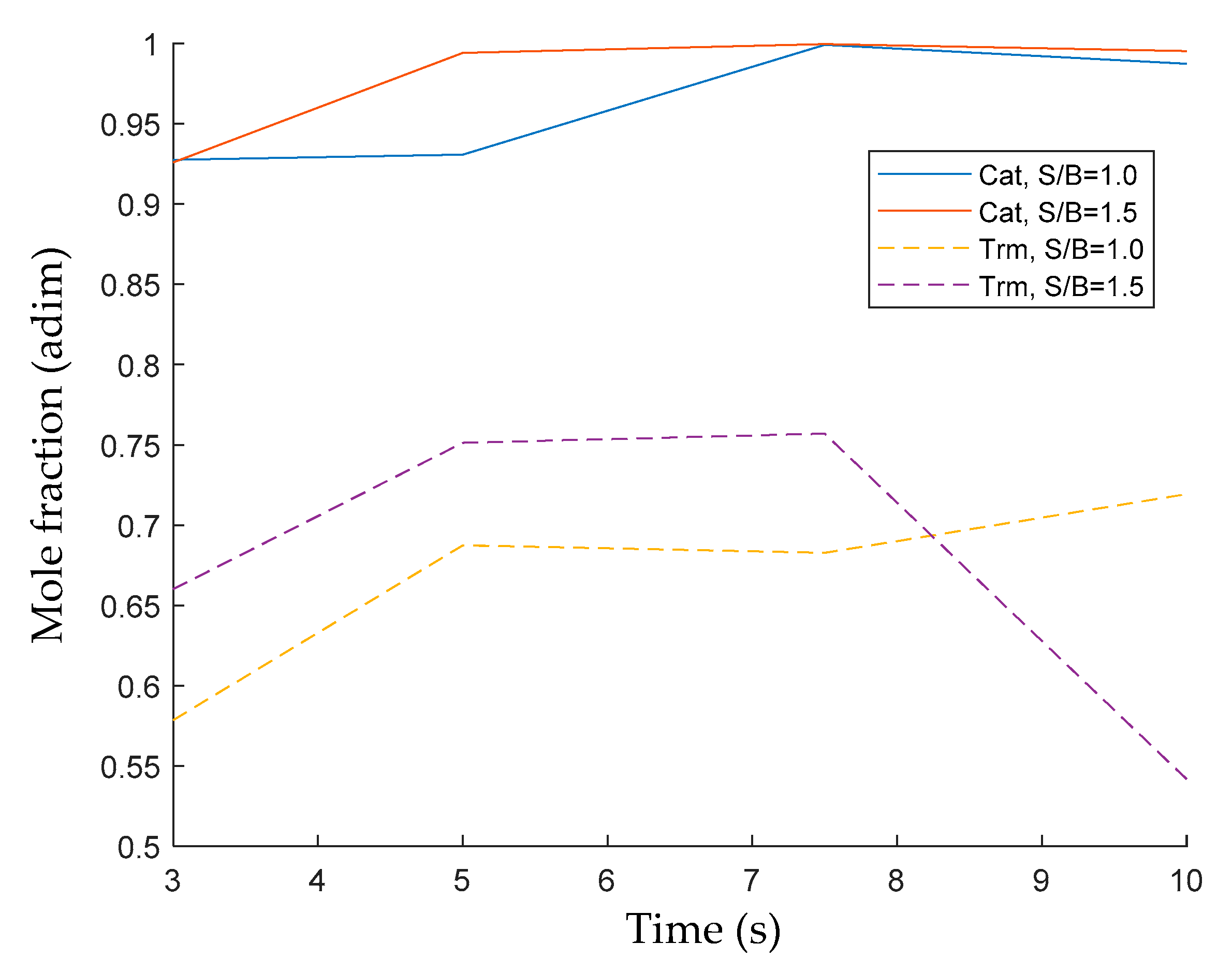
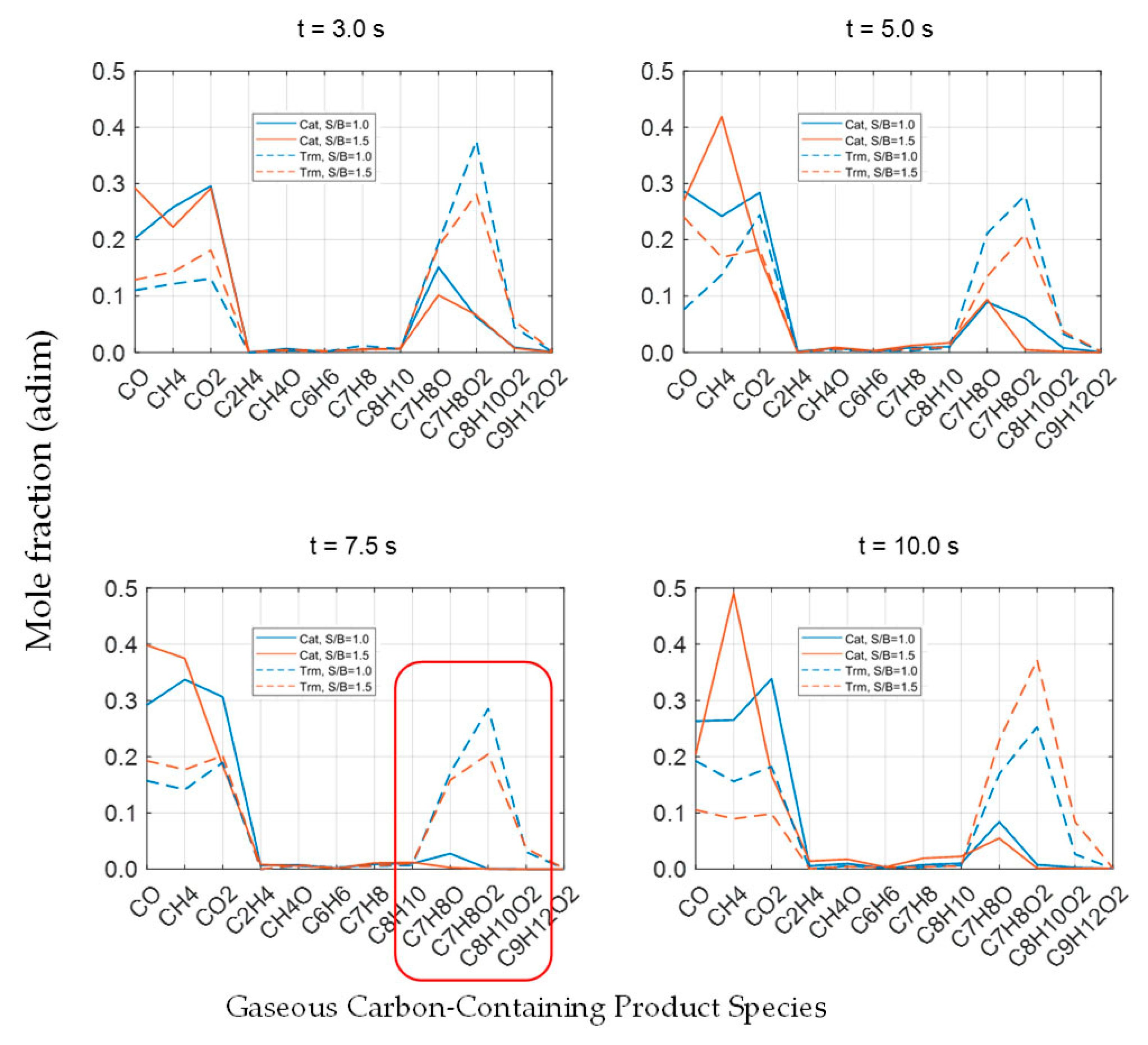
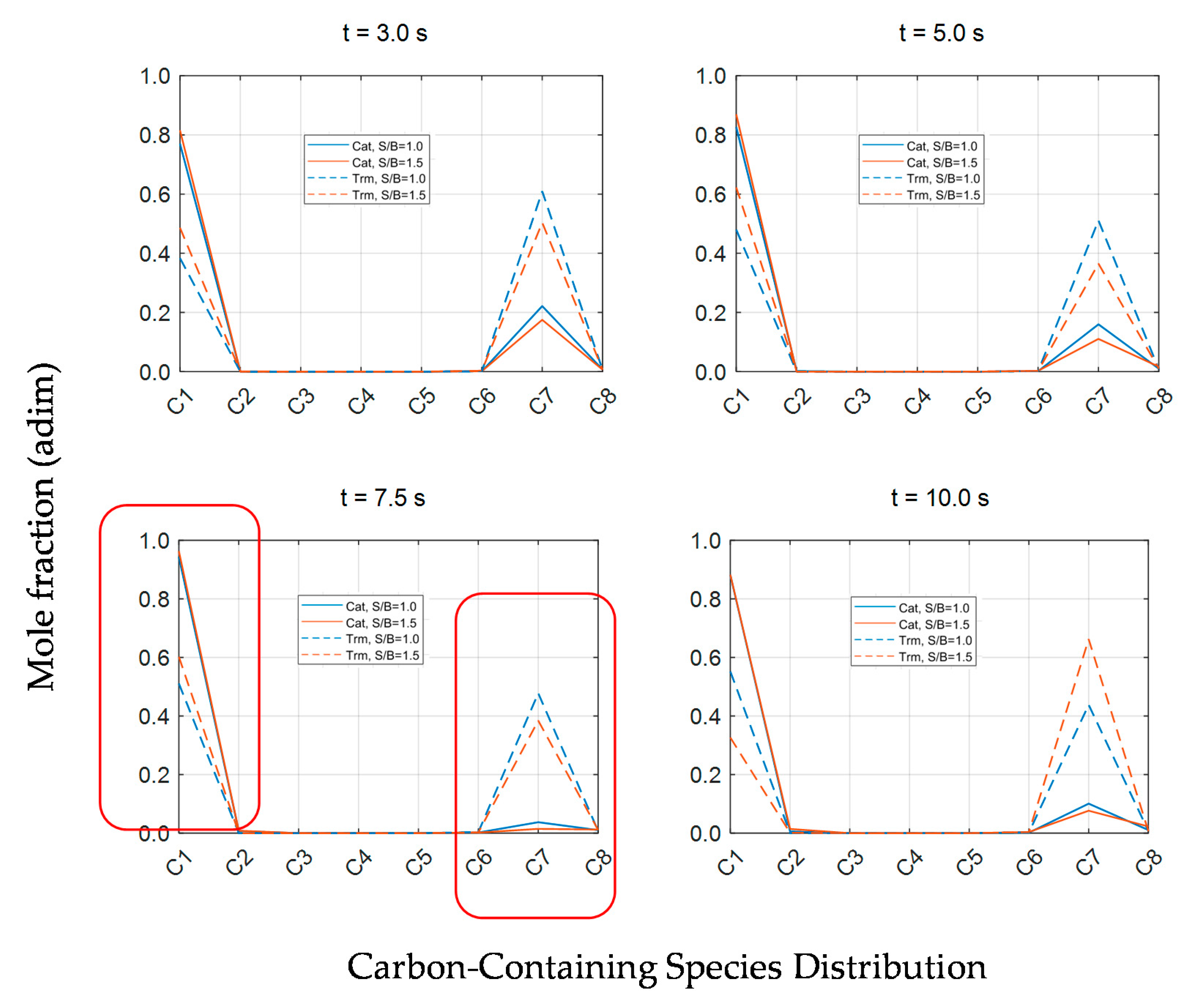
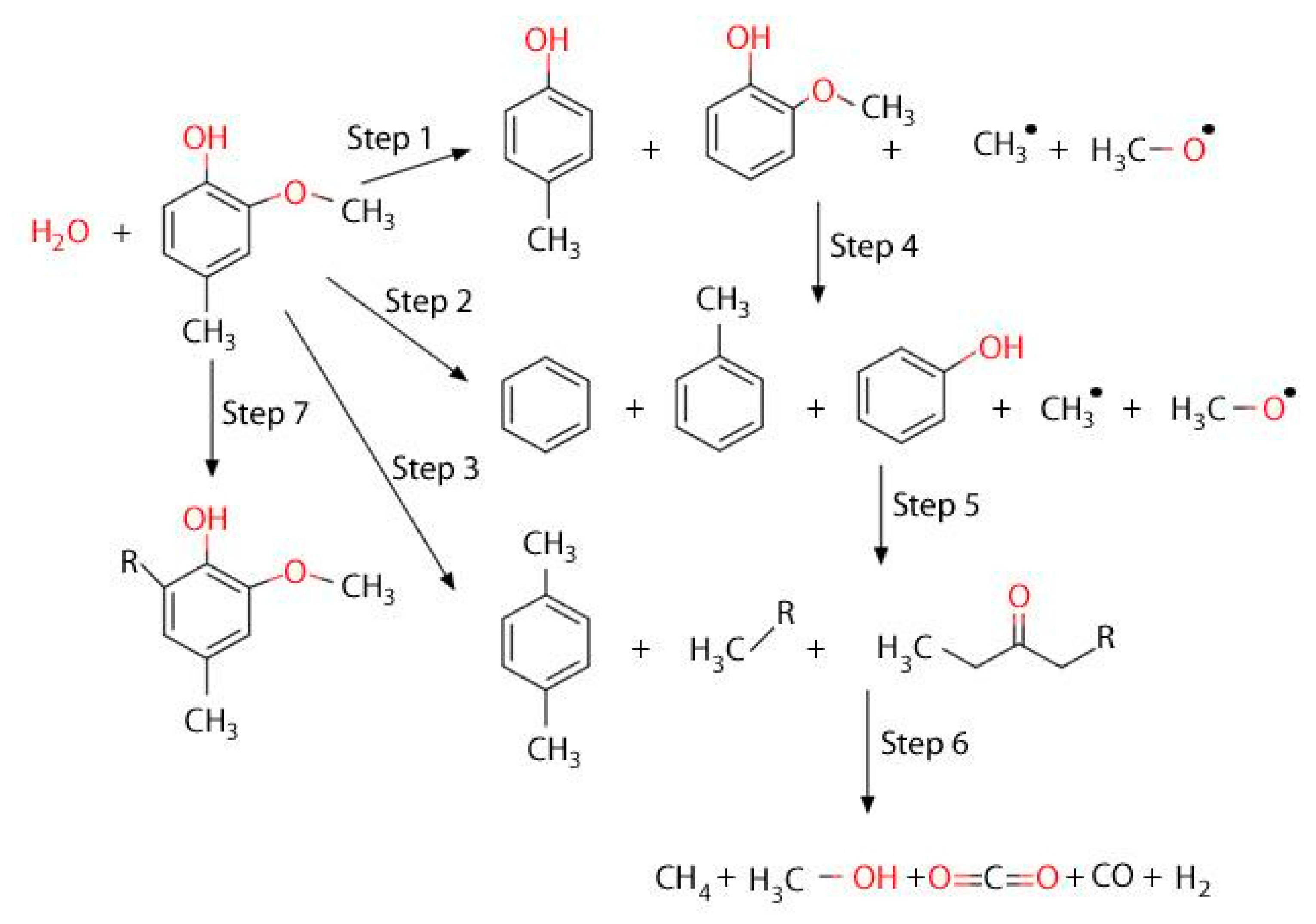
2.3.6. Evaluation of Catalytic Tar Reforming and Cracking
Conversion and Carbon Conversion Distribution
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation
3.3. Catalyst Characterization
3.3.1. N2 Adsorption
3.3.2. XRD Patterns
3.3.3. X-ray Photoelectron Spectroscopy (XPS)
3.3.4. Temperature Programmed Experiments
3.3.5. Pyridine Fourier Transform Infrared Spectroscopy (FTIR)
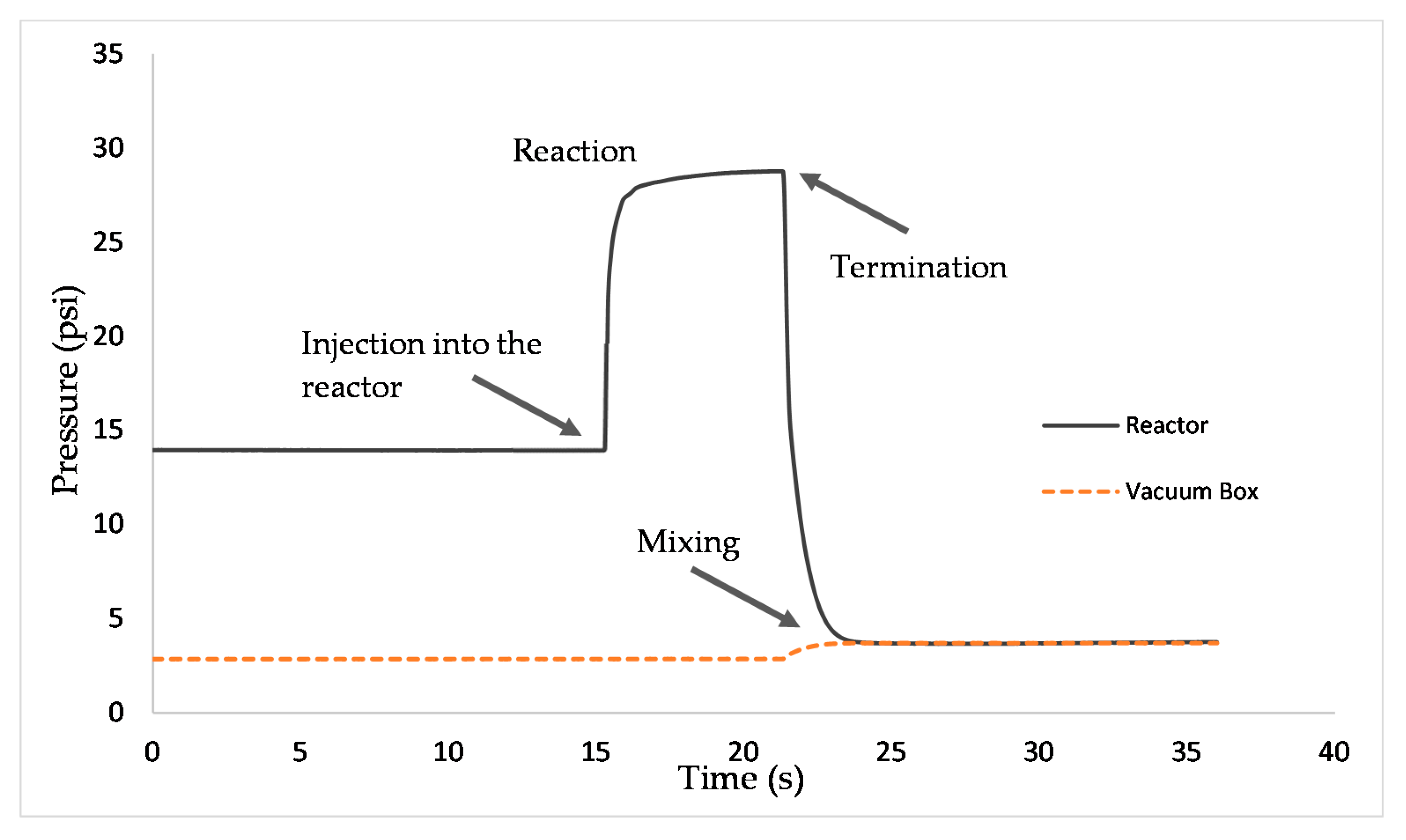
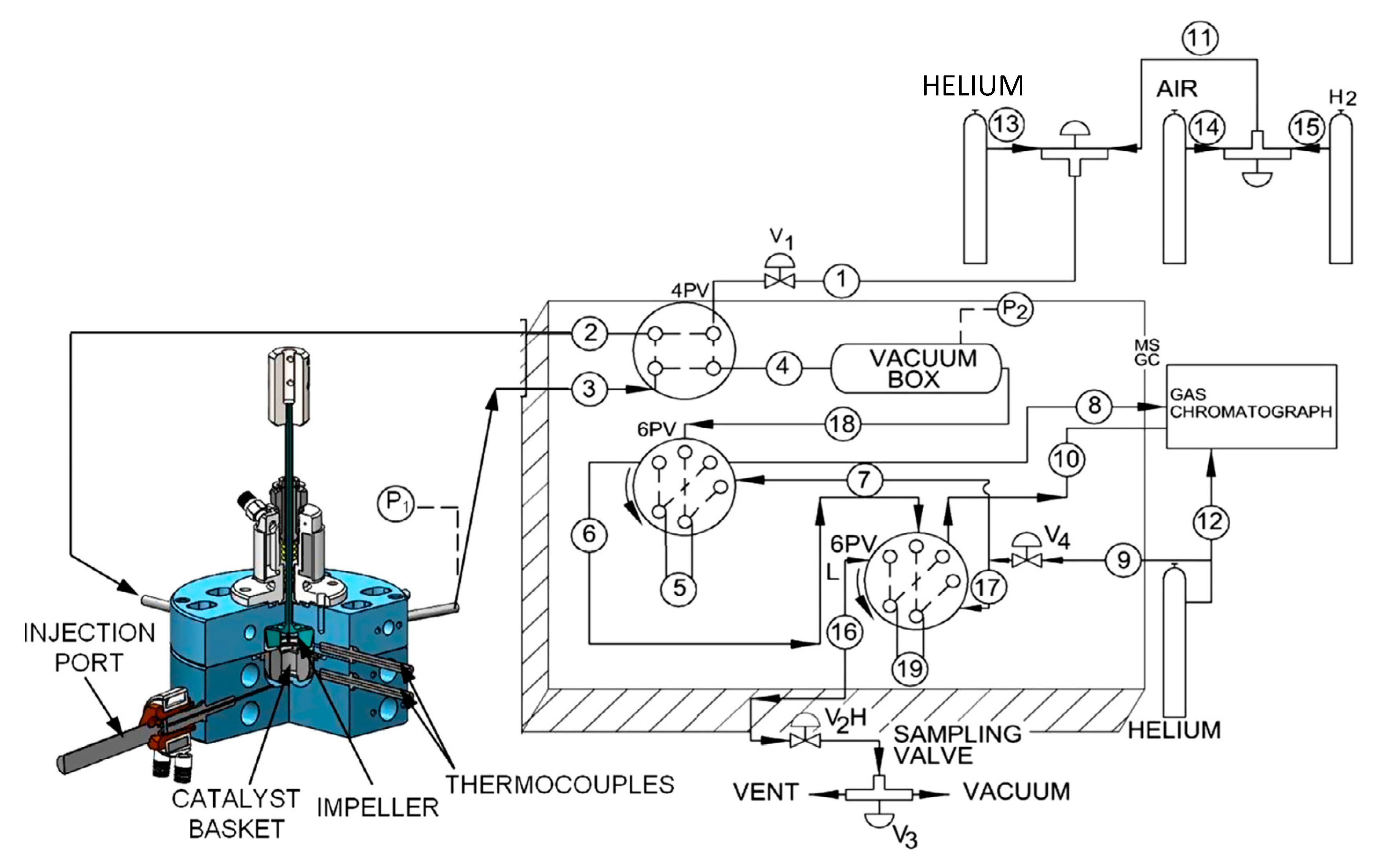
3.4. Tar Cracking Evaluation
4. Conclusions
- (a)
- A fluidizable FexOy, CaO doped, γ-Al2O3 supported catalyst, designated as FexOy/CaO-γ-Al2O3, was successfully developed. This catalyst can steam gasify methoxy-4-methylphenol, which is a model probe biomass-derived tar, with 99.95% conversion at 500 °C and S/B ratios of 1.5 and 7.5 s.
- (b)
- At these conditions, gasification yields C1-C7 species, with no significant tar remaining and 0.98% carbon deposited as coke.
- (c)
- The developed FexOy/CaO-γ-Al2O3 catalyst performs close to thermodynamic equilibrium, yielding a 96.98% C1-C2 light-fraction product selectivity.
- (d)
- This catalyst also shows excellent stability under repeated gasification and regeneration cycles, which are the expected operating conditions of a circulating fluidized bed gasifier.
- (e)
- Up to 10 wt% CaO addition helps to reduce the thermal sintering and the Lewis acidity of the γ-Al2O3 support as well as to improve its basicity. Furthermore, controlled CaO addition has a positive impact on acid-base properties, limiting pore blocking by coke.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Acronym | Descriptor |
| 2M4MP | 2-methoxy-4-methylphenol |
| BET | Brunauer–Emmett–Teller |
| XRD | X-ray diffraction |
| TPD | Temperature programmed desorption |
| TPR | Temperature programmed reduction |
| TCD | Thermal conductivity detector |
| NMR | Nuclear magnetic resonance |
| DFT | Density-functional theory |
| TPO | Temperature programmed oxidation |
| XPS | X-ray photoelectron spectroscopy |
| FWHM | Full width at half maximum |
| JCPDS | Joint Committee on Powder Diffraction Standards |
| FTIR | Fourier-transform infrared spectroscopy |
| CREC | Chemical Reactor Engineering Centre |
| TOC | Total organic carbon |
| PAHs | Polyaromatic hydrocarbons |
| STP | Standard temperature and pressure |
| PA | Pyridine proton affinity |
| CBP | Coordinated bonded pyridine |
| HBP | Hydrogen bonded pyridine |
| H | p-hydroxyphenyl |
| G | Guaiacyl |
| S | Syringyl |
| EDXRF | Energy dispersive X-ray fluorescence |
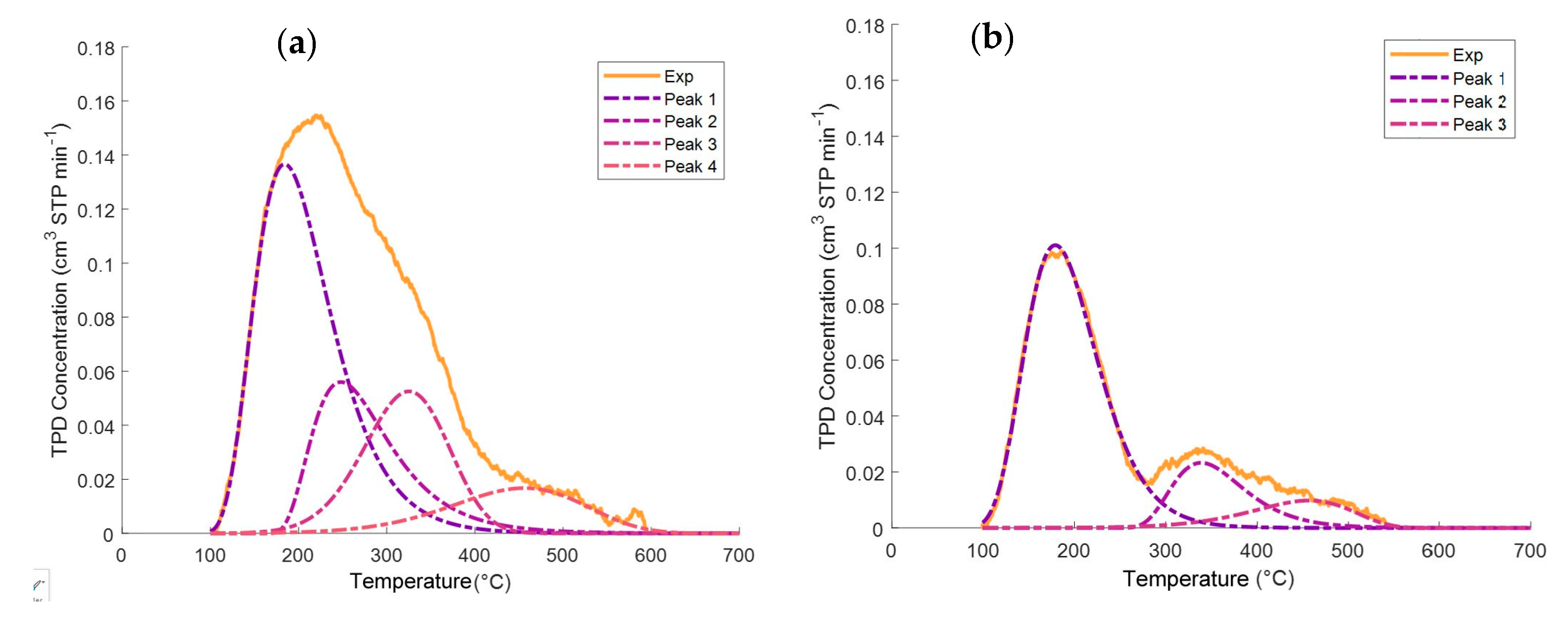
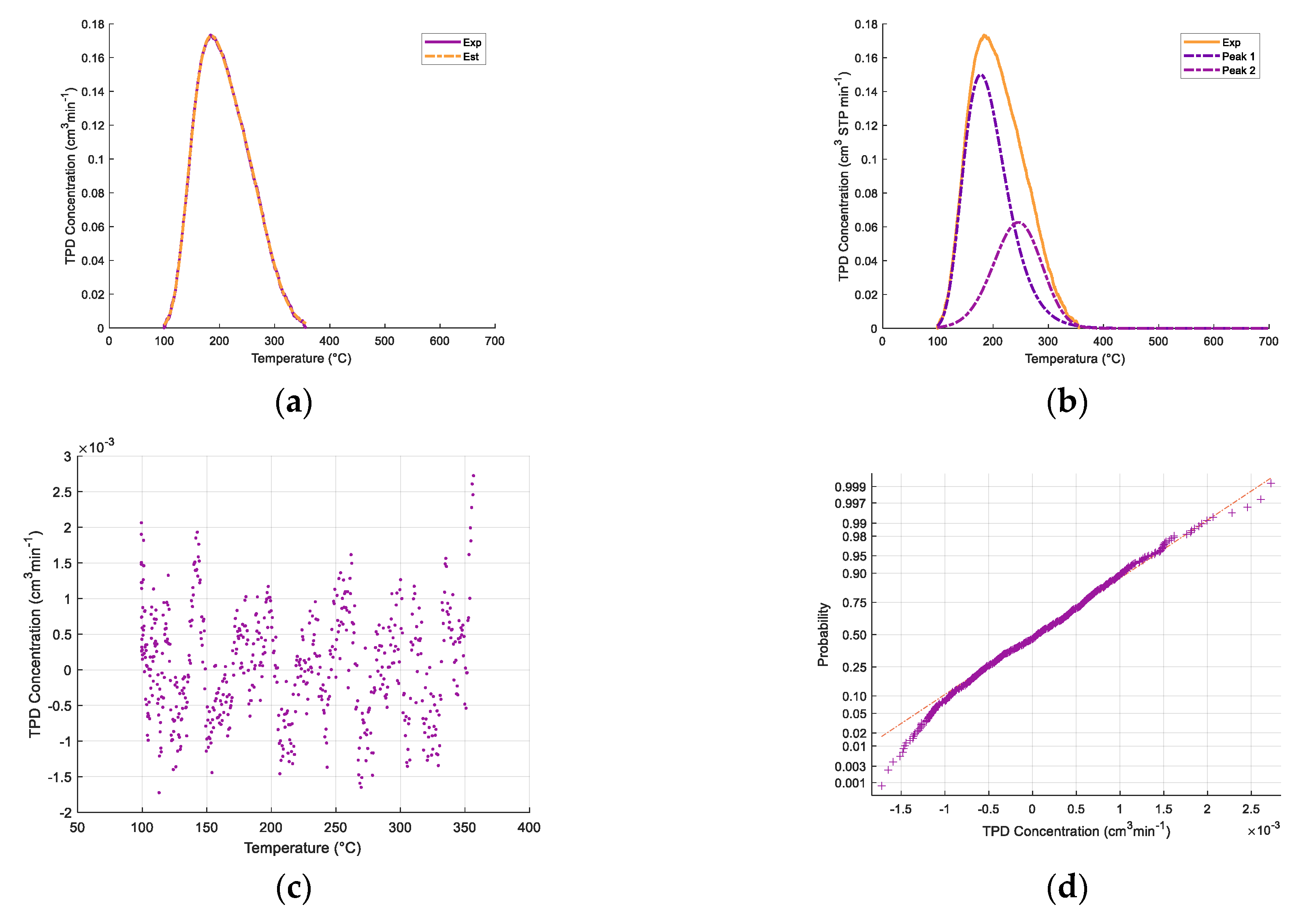
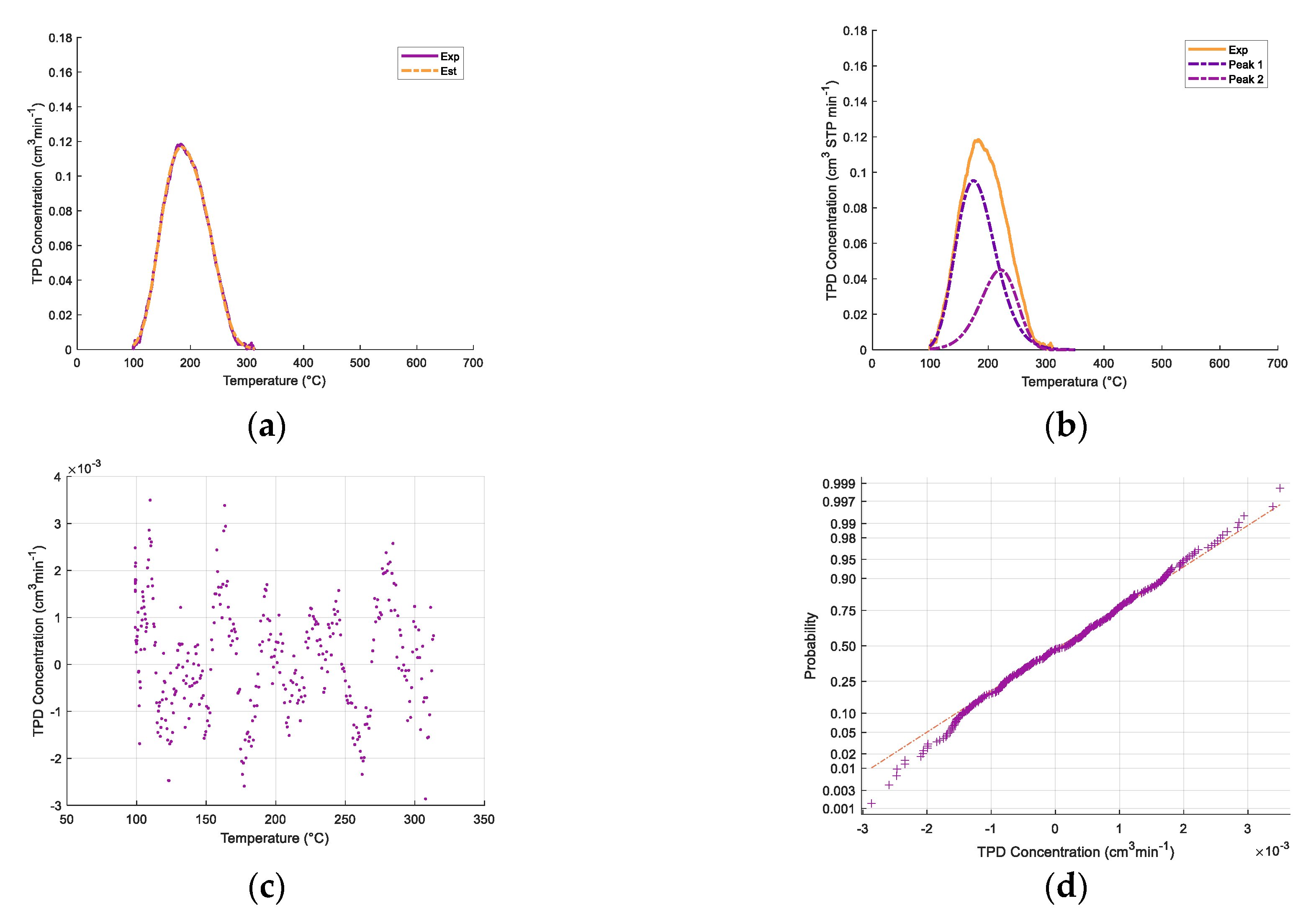
Appendix A. Deconvolution of NH3-TPD Desorption Peaks


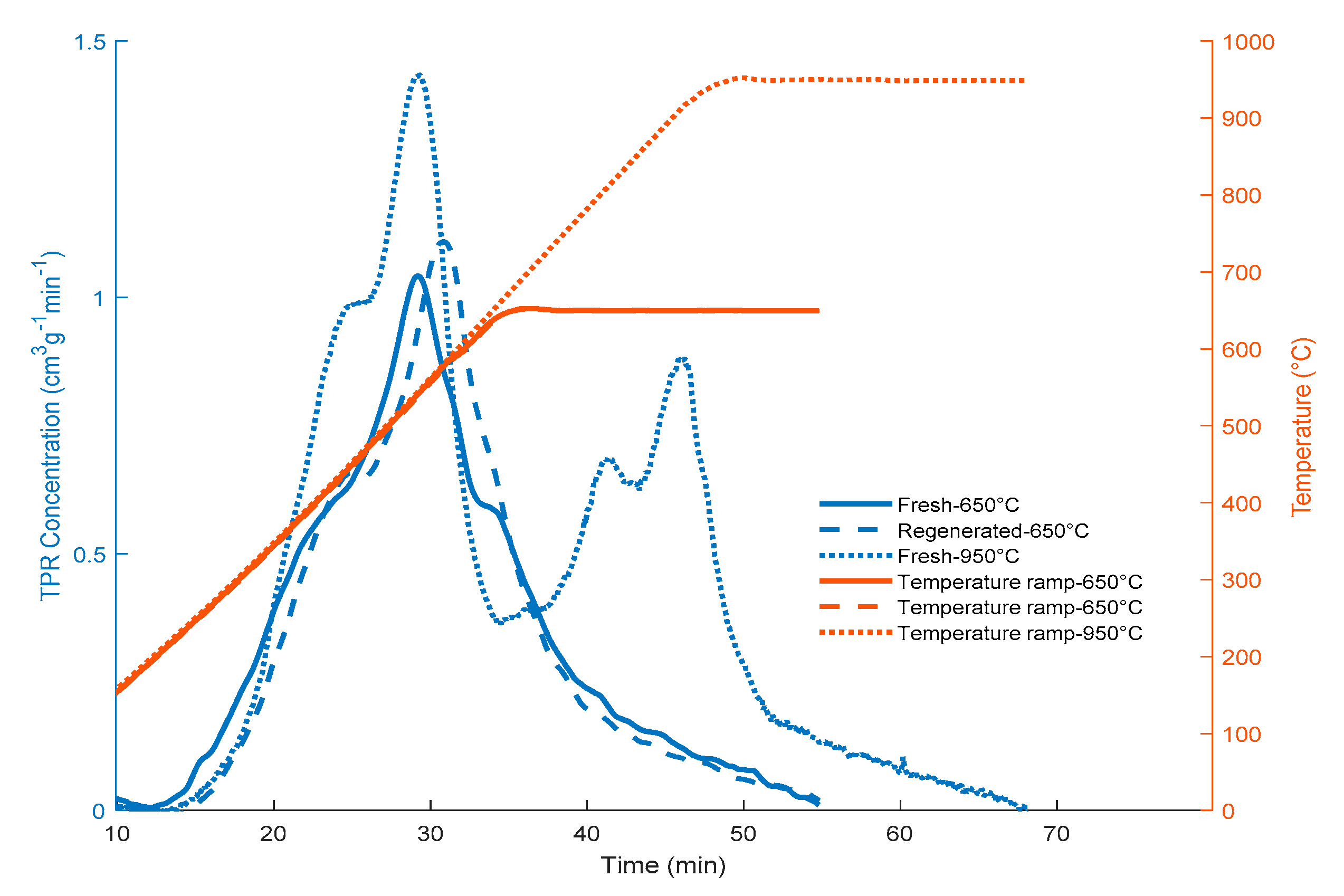
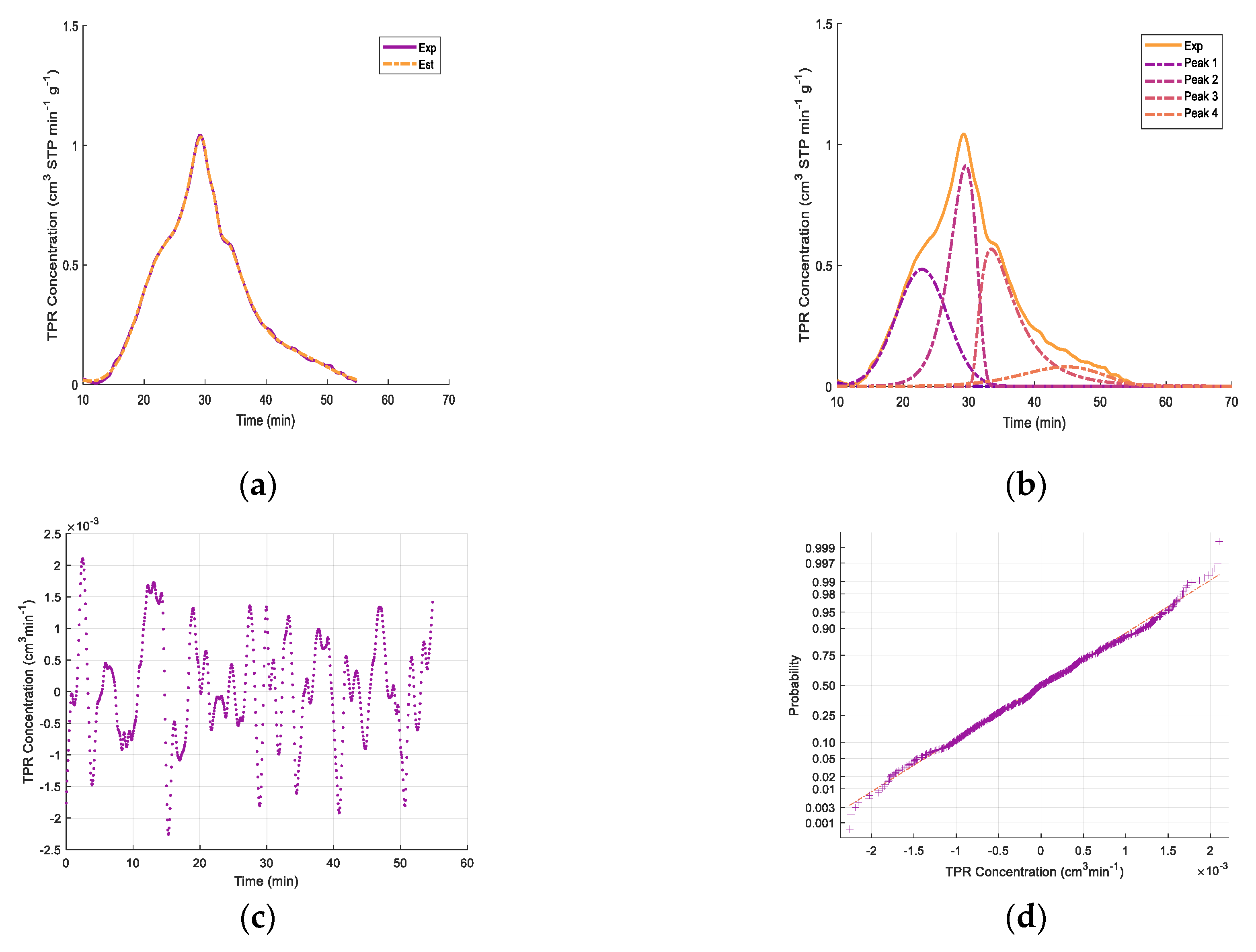
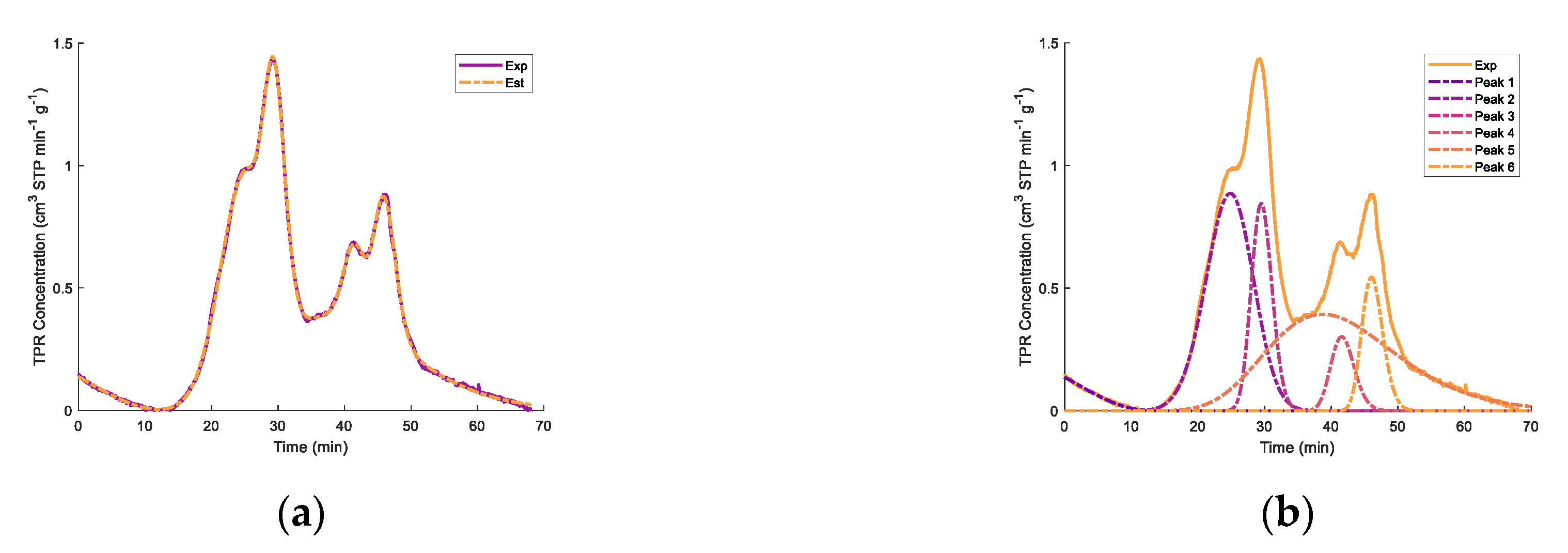
Appendix B. Deconvolution of TPR for FexOy/CaO- γ-Al2O3 Catalysts



Appendix C. Coke Deposition and Iron Evaluation

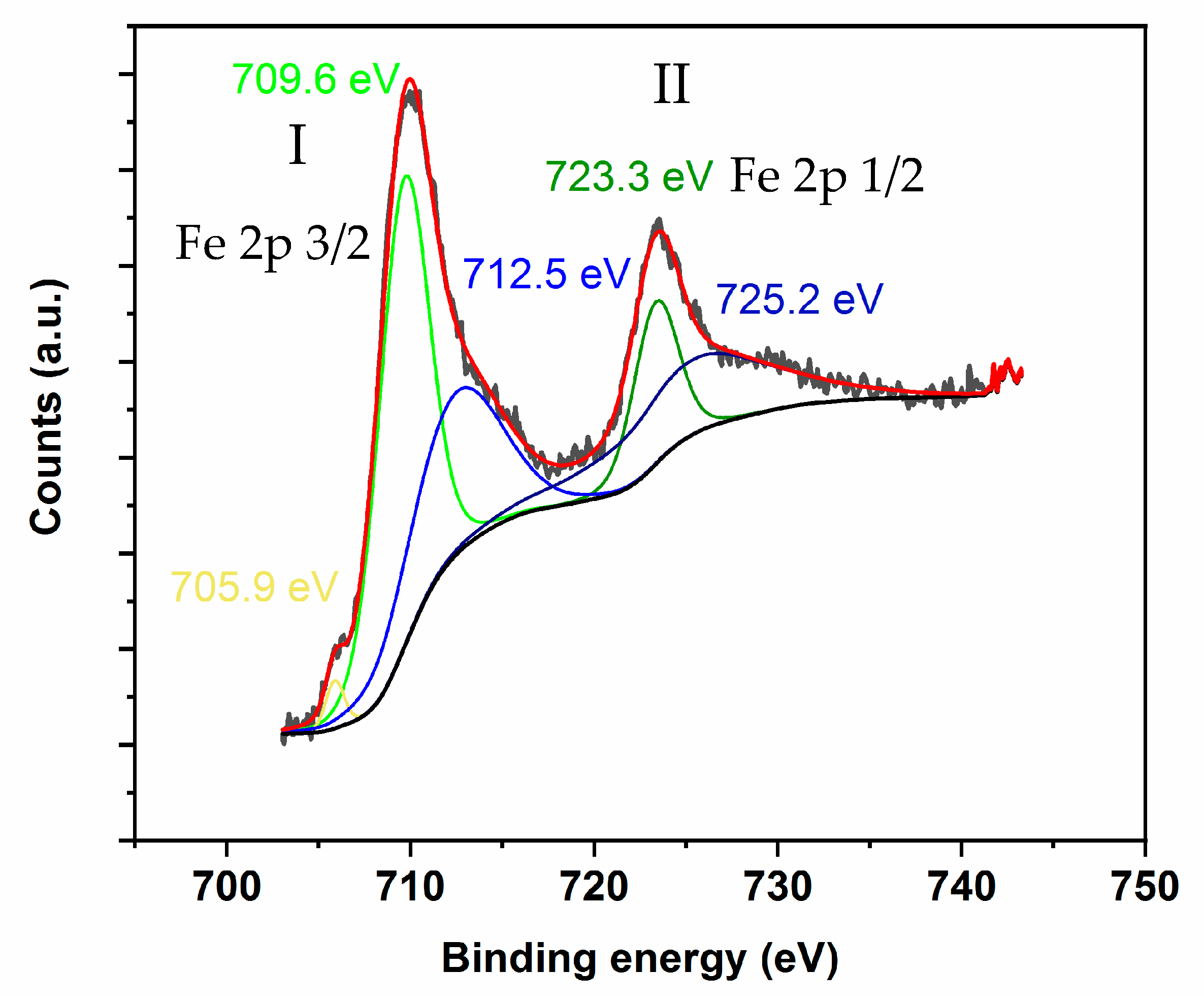
Appendix D. Evaluation of Fe0, Fe2+, and Fe3+ Distribution Using XPS and TPR Data
- (a)
- The β’ = Fe2+/Fe3+ ratio was established using XPS, as described on page 14.
- (b)
- The µ fraction = moles of H2 consumed with TPR at 650 °C/moles of H2 consumed with TPR at 950 °C was assessed as γ’ = (0.5β + 1.5α’)/1.5(1 + α’ + β’).
- (c)
- Given this, and rearranging the γ’ equation, the α’ was calculated as α = Feo/Fe3 + = (1.5γ’(1 + β’)-0.5β’)/(1.5(1-γ’)).
- (d)
- Then, on the basis of (1+ α’+ β’)100 = (1+ Fe2+/Fe3+ Feo/Fe3+) 100 or Fe3+/(Fe+ Fe2+ + Fe3+)= Fe3+%.
- (e)
- Furthermore, considering (α’ Fe3+%) and (β’ Fe3+%), both the Fe2+% and Fe0% can be calculated as well.
References
- IRENA, I. Renewable Power Generation Costs in 2017; IRENA—International Renewable Energy Agency: Abu Dhabi, UAE, 2018. [Google Scholar]
- Torres, C.; Urvina, L.; Hernández, N.; Molina, D. Informe Técnico Evaluación de Desempeño de la Tecnología Comercial de Gasificación Utilizando Residuos de Café; Universidad de Costa Rica: San José, Costa Rica, 2016. [Google Scholar]
- De Lasa, H.; Salaices, E.; Mazumder, J.; Lucky, R. Catalytic steam gasification of biomass: Catalysts, thermodynamics and kinetics. Chem. Rev. 2011, 111, 5404–5433. [Google Scholar] [CrossRef]
- Tanksale, A.; Beltramini, J.N.; Lu, G.M. A review of catalytic hydrogen production processes from biomass. Renew. Sustain. Energy Rev. 2010, 14, 166–182. [Google Scholar] [CrossRef]
- Chan, F.L.; Tanksale, A. Review of recent developments in Ni-based catalysts for biomass gasification. Renew. Sustain. Energy Rev. 2014, 38, 428–438. [Google Scholar] [CrossRef]
- Chianese, S.; Fail, S.; Binder, M.; Rauch, R.; Hofbauer, H.; Molino, A.; Blasi, A.; Musmarra, D. Experimental investigations of hydrogen production from CO catalytic conversion of tar rich syngas by biomass gasification. Catal. Today 2016, 277, 182–191. [Google Scholar] [CrossRef]
- Li, S.; Zheng, H.; Zheng, Y.; Tian, J.; Jing, T.; Chang, J.-S.; Ho, S.-H. Recent advances in hydrogen production by thermo-catalytic conversion of biomass. Int. J. Hydrogen Energy 2019, 44, 14266–14278. [Google Scholar] [CrossRef]
- Li, C.; Suzuki, K. Tar property, analysis, reforming mechanism and model for biomass gasification-An overview. Renew. Sustain. Energy Rev. 2009, 13, 594–604. [Google Scholar] [CrossRef]
- Li, D.; Tamura, M.; Nakagawa, Y.; Tomishige, K. Metal catalysts for steam reforming of tar derived from the gasification of lignocellulosic biomass. Bioresour. Technol. 2015, 178, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.J.; Milne, T.A. Chemistry of Tar Formation and Maturation in the Thermochemical Conversion of Biomass. In Developments in Thermochemical Biomass Conversion; Springer: Dordrecht, The Netherlands, 1997; pp. 803–816. [Google Scholar]
- Abatzoglou, N.; Barker, N.; Hasler, P.; Knoef, H. The development of a draft protocol for the sampling and analysis of particulate and organic contaminants in the gas from small biomass gasifiers. Biomass Bioenergy 2000, 18, 5–17. [Google Scholar] [CrossRef]
- Maniatis, K.; Beenackers, A.A.C.M. Tar Protocols. IEA Bioenergy Gasification Task: Introduction. Biomass Bioenergy 2000, 18, 1–4. [Google Scholar] [CrossRef]
- Woolcock, P.J.; Brown, R.C. A review of cleaning technologies for biomass-derived syngas. Biomass Bioenergy 2013, 52, 54–84. [Google Scholar] [CrossRef]
- Rakesh, N.; Dasappa, S. A critical assessment of tar generated during biomass gasification—Formation, evaluation, issues and mitigation strategies. Renew. Sustain. Energy Rev. 2018, 91, 1045–1064. [Google Scholar] [CrossRef]
- Asadullah, M. Barriers of commercial power generation using biomass gasification gas: A review. Renew. Sustain. Energy Rev. 2014, 29, 201–215. [Google Scholar] [CrossRef]
- Anis, S.; Zainal, Z.A. Tar reduction in biomass producer gas via mechanical, catalytic and thermal methods: A review. Renew. Sustain. Energy Rev. 2011, 15, 2355–2377. [Google Scholar] [CrossRef]
- Mozaffarian, M.; Cameron, L.R.; Falzon, J.P.J.; Cohen, B.; Das, K.C.; Bole-Rentel, T. Biomass Waste-to-Energy Toolkit for Development Practitioners The Green House SNV Netherlands Development Organisation; ECN: Petten, The Netherlands, 2014. [Google Scholar]
- Sikarwar, V.S.; Zhao, M.; Clough, P.; Yao, J.; Zhong, X.; Memon, M.Z.; Fennell, P.S. An overview of advances in biomass gasification. Energy Environ. Sci. 2016, 9, 2939–2977. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Wu, S.; Wu, Y.; Gao, J. Structure characteristics and gasification activity of residual carbon from updraft fixed-bed biomass gasification ash. Energy Convers Manag. 2017, 136, 108–118. [Google Scholar] [CrossRef]
- Pissot, S.; Vilches, T.B.; Thunman, H.; Seemann, M. Biomass and Bioenergy Effect of ash circulation on the performance of a dual fluidized bed gasification system. Biomass Bioenergy 2018, 115, 45–55. [Google Scholar] [CrossRef]
- Shahbaz, M.; Yusup, S.; Inayat, A.; Patrick, D.O.; Ammar, M. The influence of catalysts in biomass steam gasification and catalytic potential of coal bottom ash in biomass steam gasification: A review. Renew. Sustain. Energy Rev. 2017, 73, 468–476. [Google Scholar] [CrossRef]
- Heinze, T. Cellulose Chemistry and Properties: Fibers Nanocelluloses and Advanced Materials; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Herman, A.P.; Yusup, S.; Shahbaz, M. Utilization of bottom ash as catalyst in biomass steam gasification for hydrogen and syngas production. Chem. Eng. Trans. 2016, 52, 1249–1254. [Google Scholar]
- Kryca, J.; Pri, J.; Joanna, Ł.; Kuba, M.; Hofbauer, H. Apparent kinetics of the water-gas-shift reaction in biomass gasi fi cation using ash-layered olivine as catalyst. Chem. Eng. J. 2018, 346, 113–119. [Google Scholar] [CrossRef]
- Boström, D.; Skoglund, N.; Grimm, A.; Boman, C.; Ohman, M.; Brostrom, M.; Backman, R. Ash transformation chemistry during combustion of biomass. Energy Fuels 2012, 26, 85–93. [Google Scholar] [CrossRef]
- Wang, L.; Skjevrak, G.; Hustad, J.E.; Skreiberg, Ø. Investigation of biomass ash sintering characteristics and the effect of additives. Energy Fuels 2014, 28, 208–218. [Google Scholar] [CrossRef]
- Sivula, L.; Oikari, A.; Rintala, J. Toxicity of waste gasification bottom ash leachate. Waste Manag. 2012, 32, 1171–1178. [Google Scholar] [CrossRef]
- Chen, Z.; Dun, Q.; Shi, Y.; Lai, D.; Zhou, Y.; Gao, S.; Xu, G. High quality syngas production from catalytic coal gasification using disposable Ca(OH)2catalyst. Chem. Eng. J. 2017, 316, 842–849. [Google Scholar] [CrossRef]
- Xu, C.; Chen, S.; Soomro, A.; Sun, Z.; Xiang, W. Hydrogen rich syngas production from biomass gasification using synthesized Fe/CaO active catalysts. J. Energy Inst. 2018, 91, 805–816. [Google Scholar] [CrossRef]
- Yin, F.; Tremain, P.; Yu, J.; Doroodchi, E.; Moghtaderi, B. Investigations on the synergistic effects of oxygen and CaO for biotars cracking during biomass gasification. Energy Fuels 2017, 31, 587–598. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, Z.; Tang, A.; Huang, H.; Wei, D.; Yu, E.; Lu, W. Steam-gasification of biomass with CaO as catalyst for hydrogen-rich syngas production. J. Energy Inst. 2019, 92, 1641–1646. [Google Scholar] [CrossRef]
- Zamboni, I.; Zimmermann, Y.; Kiennemann, A.; Courson, C. Improvement of steam reforming of toluene by CO2 capture using Fe/CaO-Ca12Al14O33 bi-functional materials. Int. J. Hydrogen Energy 2015, 40, 5297–5304. [Google Scholar] [CrossRef]
- Kuba, M.; Kirnbauer, F.; Hofbauer, H. Influence of coated olivine on the conversion of intermediate products from decomposition of biomass tars during gasification. Biomass Convers. Biorefinery 2017, 7, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sun, Z.; Zhang, Q.; Hu, J.; Xiang, W. Biomass and Bioenergy Steam gasi fi cation of sewage sludge with CaO as CO 2 sorbent for hydrogen-rich syngas production. Biomass Bioenergy 2017, 107, 52–62. [Google Scholar] [CrossRef]
- Abu El-Rub, Z.; Bramer, E.A.; Brem, G. Review of Catalysts for Tar Elimination in Biomass Gasification. Ind. Eng. Chem. Res. 2004, 43, 6911–6919. [Google Scholar] [CrossRef]
- Rapagnà, S.; Virginie, M.; Gallucci, K.; Courson, C.; Di Marcello, M.; Kiennemann, A.; Foscolo, P.U. Fe/olivine catalyst for biomass steam gasification: Preparation, characterization and testing at real process conditions. Catal. Today 2011, 176, 163–168. [Google Scholar] [CrossRef]
- Zamboni, I.; Courson, C.; Kiennemann, A. Fe-Ca interactions in Fe-based/CaO catalyst/sorbent for CO2 sorption and hydrogen production from toluene steam reforming. Appl. Catal. B Environ. 2017, 203, 154–165. [Google Scholar] [CrossRef]
- Devi, L.; Craje, M.; Thüne, P.; Ptasinski, K.J.; Janssen, F.J. Olivine as tar removal catalyst for biomass gasifiers: Catalyst characterization. Appl. Catal. A Gen. 2005, 294, 68–79. [Google Scholar] [CrossRef]
- Fredriksson, H.O.; Lancee, R.J.; Thüne, P.C.; Veringa, H.J.; Niemantsverdriet, J.H. Olivine as tar removal catalyst in biomass gasification: Catalyst dynamics under model conditions. Appl. Catal. B Environ. 2013, 130, 168–177. [Google Scholar] [CrossRef]
- Nordgreen, T. Iron-Based Materials as Tar Cracking Catalyst in Waste Gasification; KTH-Royal Institute of Technology: Stockholm, Sweden, 2011. [Google Scholar]
- Kastner, J.R.; Mani, S.; Juneja, A. Catalytic decomposition of tar using iron supported biochar. Fuel Process. Technol. 2015, 130, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Adnan, M.A.; Muraza, O.; Razzak, S.A.; Hossain, M.M.; de Lasa, H.I. Iron Oxide over Silica-Doped Alumina Catalyst for Catalytic Steam Reforming of Toluene as a Surrogate Tar Biomass Species. Energy Fuels 2017, 31, 7471–7481. [Google Scholar] [CrossRef]
- Di Felice, L.; Courson, C.; Foscolo, P.U.; Kiennemann, A. Iron and nickel doped alkaline-earth catalysts for biomass gasification with simultaneous tar reformation and CO2 capture. Int. J. Hydrogen Energy 2011, 36, 5296–5310. [Google Scholar] [CrossRef]
- Bastos, A.K.; Torres, C.; Mazumder, A.; de Lasa, H. CO2 biomass fluidized gasification: Thermodynamics and reactivity studies. Can. J. Chem. Eng. 2018, 96, 2176–2184. [Google Scholar] [CrossRef]
- Mitsuoka, K.; Hayashi, S.; Amano, H.; Kayahara, K.; Sasaoaka, E.; Uddin, M.A. Gasification of woody biomass char with CO2: The catalytic effects of K and Ca species on char gasification reactivity. Fuel Process. Technol. 2011, 92, 26–31. [Google Scholar] [CrossRef]
- Perander, M.; DeMartini, N.; Brink, A.; Kramb, J.; Karlström, O.; Hemming, J.; Hupa, M. Catalytic effect of Ca and K on CO2 gasification of spruce wood char. Fuel 2015, 150, 464–472. [Google Scholar] [CrossRef]
- Kajita, M.; Kimura, T.; Norinaga, K.; Li, C.Z.; Hayashi, J.I. Catalytic and noncatalytic mechanisms in steam gasification of char from the pyrolysis of biomass. Energy Fuels 2010, 24, 108–116. [Google Scholar] [CrossRef]
- Abdoulmoumine, N.; Adhikari, S.; Kulkarni, A.; Chattanathan, S. A review on biomass gasification syngas cleanup. Appl. Energy 2015, 155, 294–307. [Google Scholar] [CrossRef]
- Liu, B.; He, Q.; Jiang, Z.; Xu, R.; Hu, B. Relationship between coal ash composition and ash fusion temperatures. Fuel 2013, 105, 293–300. [Google Scholar] [CrossRef]
- Jiang, M.Q.; Zhou, R.; Hu, J.; Wang, F.C.; Wang, J. Calcium-promoted catalytic activity of potassium carbonate for steam gasification of coal char: Influences of calcium species. Fuel 2012, 99, 64–71. [Google Scholar] [CrossRef]
- Teixeira, P.; Lopes, H.; Gulyurtlu, I.; Lapa, N.; Abelha, P. Evaluation of slagging and fouling tendency during biomass co-firing with coal in a fluidized bed. Biomass Bioenergy 2012, 39, 192–203. [Google Scholar] [CrossRef] [Green Version]
- Piotrowska, P.; Zevenhoven, M.; Davidsson, K.; Hupa, M.; Åmand, L.E.; Barišić, V.; Coda Zabetta, E. Fate of alkali metals and phosphorus of rapeseed cake in circulating fluidized bed boiler part 1: Cocombustion with wood. Energy Fuels 2010, 24, 333–345. [Google Scholar] [CrossRef]
- Ropp, R.C. Inorganic Polymeric Glasses; Elsevier: Amsterdam, The Netherlands, 2013; Volume 15. [Google Scholar]
- Greaves, G.N.; Sen, S. Inorganic glasses, glass-forming liquids and amorphizing solids. Adv. Phys. 2007, 56, 1–166. [Google Scholar] [CrossRef]
- Mazumder, J.; De Lasa, H. Fluidizable Ni/La2O3-γ-Al2O3 catalyst for steam gasification of a cellulosic biomass surrogate. Appl. Catal. B Environ. 2014, 160, 67–79. [Google Scholar] [CrossRef]
- Argyle, M.; Bartholomew, C. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef] [Green Version]
- Slater, A.G.; Cooper, A.I. Function-led design of new porous materials. Science 2015, 348, aaa8075. [Google Scholar] [CrossRef]
- Leng, Y. Materials Characterization: Introduction to Microscopic and Spectroscopic Methods; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Waseda, Y.; Matsubara, E.; Shinoda, K. X-ray Diffraction Crystallography: Introduction, Examples and Solved Problems; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Kashiwaya, Y. Crystal Phases Formed in a CaO-Fe2O3 System Under a High Cooling Rate in Air. Metall. Mater. Trans. B 2017, 48, 3228–3238. [Google Scholar] [CrossRef]
- Védrine, J.C. Acid-base characterization of heterogeneous catalysts: An up-to-date overview. Res. Chem. Intermed. 2015, 41, 9387–9423. [Google Scholar] [CrossRef]
- Peri, J.B. A model for the surface of γ-alumin. J. Phys. Chem. 1965, 69, 220–230. [Google Scholar] [CrossRef]
- Peri, J.B. Infrared and Gravimetric Study of the Surface Hydration of 7-Alumina. J. Phys. Chem. 1965, 69, 211–219. [Google Scholar] [CrossRef]
- Peri, J.B. Infrared study of adsorption of carbon dioxide, hydrogen chloride, and other molecules on acid sites on dry silica-alumina and -γ-alumina 1. J. Phys. Chem. 1966, 70, 3168–3179. [Google Scholar] [CrossRef]
- Dewing, J.; Monks, G.T.; Youll, B. Competitive adsorption of pyridine and sterically hindered pyridines on alumina. J. Catal. 1976, 44, 226–235. [Google Scholar] [CrossRef]
- Digne, M.; Sautet, P.; Raybaud, P.; Euzen, P.; Toulhoat, H. Use of DFT to achieve a rational understanding of acid-basic properties of γ-alumina surfaces. J. Catal. 2004, 226, 54–68. [Google Scholar] [CrossRef]
- Majors, P.D.; Raidy, T.E.; Ellis, P.D. A Multinuclear Solid-State NMR Investigation of the Chemisorption of Ammonia on 7-Alumina. J. Am. Chem. Soc. 1986, 108, 8123–8129. [Google Scholar] [CrossRef]
- Majors, P.D.; Ellis, P.D. Surface Site Distributions by Solid-State Multinuclear NMR Spectroscopy. Pyridine Binding to 7-Alumina by 15N and 2H. J. Am. Chem. Soc. 1987, 109, 1648–1653. [Google Scholar] [CrossRef]
- Coster, D.; Blumenfeld, A.L.; Fripiat, J.J. Lewis acid sites and surface aluminum in aluminas and zeolites: A high-resolution NMR study. J. Phys. Chem. 1994, 98, 6201–6211. [Google Scholar] [CrossRef]
- Wischert, R.; Copéret, C.; Delbecq, F.; Sautet, P. Optimal Water Coverage on Alumina: A Key to Generate Lewis Acid-Base Pairs that are Reactive Towards the C-H Bond Activation of Methane. Angew. Chemie 2011, 123, 3260–3263. [Google Scholar] [CrossRef]
- Morterra, C.; Magnacca, G. A case study: Surface chemistry and surface structure of catalytic aluminas, as studied by vibrational spectroscopy of adsorbed species. Catal. Today 1996, 27, 497–532. [Google Scholar] [CrossRef]
- Lagauche, M.; Larmier, K.; Jolimaitre, E.; Barthelet, K.; Chizallet, C. Thermodynamic Characterization of the Hydroxyls Group on the γ -Alumina Surface by the Energy Distribution Function. J. Phys. Chem. C 2017, 121, 16770–16782. [Google Scholar] [CrossRef]
- Samain, L.; Jaworski, A.; Edén, M.; Ladd, D.M.; Seo, D.K.; Garcia-Garcia, F.J.; Häussermann, U. Structural analysis of highly porous γ-Al2O3. J. Solid State Chem. 2014, 217, 1–8. [Google Scholar] [CrossRef]
- Scroder, K.; Junge, B.; Bitterlich, M. Topics in Organometallic Chemistry—Iron Catalysis; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1–3. [Google Scholar]
- Bauer, E.B. Iron catalysis: Historic overview and current trends. Top. Organomet. Chem. 2015, 50, 259–310. [Google Scholar]
- Padrón, J.I.; Martin, V.S. Catalysis by Means of Fe-Based Lewis Acids. In Iron Catalysis; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1–26. [Google Scholar]
- Parry, E.P. An infrared study of pyridine adsorbed on acidic solids. Characterization of surface acidity. J. Catal. 1963, 2, 371–379. [Google Scholar] [CrossRef]
- Lee, M.H.; Cheng, C.F.; Heine, V.; Klinowski, J. Distribution of tetrahedral and octahedral Al sites in gamma alumina. Chem. Phys. Lett. 1997, 265, 673–676. [Google Scholar] [CrossRef]
- Huggins, B.A.; Ellis, P.D. 27A1 Nuclear Magnetic Resonance Study of Aluminas and Their Surfaces. J. Am. Chem. Soc. 1992, 114, 2098–2108. [Google Scholar] [CrossRef]
- Liu, X. DRIFTS study of surface of γ-alumina and its dehydroxylation. J. Phys. Chem. C 2008, 112, 5066–5073. [Google Scholar] [CrossRef]
- Mazumder, J.; de Lasa, H.I. Fluidizable La2O3promoted Ni/γ-Al2O3 catalyst for steam gasification of biomass: Effect of catalyst preparation conditions. Appl. Catal. B Environ. 2015, 168–169, 250–265. [Google Scholar] [CrossRef]
- Pineau, A.; Kanari, N.; Gaballah, I. Kinetics of reduction of iron oxides by H2. Part I: Low temperature reduction of hematite. Thermochim. Acta 2006, 447, 89–100. [Google Scholar] [CrossRef]
- Pineau, A.; Kanari, N.; Gaballah, I. Kinetics of reduction of iron oxides by H2. Part II. Low temperature reduction of magnetite. Thermochim. Acta 2007, 456, 75–88. [Google Scholar] [CrossRef]
- Lin, H.; Chen, Y.; Li, C. The mechanism of reduction of iron oxide by hydrogen. Thermochim. Acta 2003, 400, 61–67. [Google Scholar] [CrossRef]
- Jeong, M.H.; Lee, D.H.; Bae, J.W. Reduction and oxidation kinetics of different phases of iron oxides. Int. J. Hydrog. Energy 2015, 40, 2613–2620. [Google Scholar] [CrossRef]
- Wei, X.; Zhou, Y.; Li, Y.; Shen, W. Polymorphous transformation of rod-shaped iron oxides and their catalytic properties in selective reduction of NO by NH3. Royal Society of Chemistry. 2015, 66141–66146. [Google Scholar] [CrossRef]
- Duman, G.; Watanabe, T.; Uddin, M.A.; Yanik, J. Steam gasification of safflower seed cake and catalytic tar decomposition over ceria modified iron oxide catalysts. Fuel Process. Technol. 2014, 126, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Bleeker, M.F.; Veringa, H.J.; Kersten, S.R.A. Pure hydrogen production from pyrolysis oil using the steam-iron process: Effects of temperature and iron oxide conversion in the reduction. Ind. Eng. Chem. Res. 2010, 49, 53–64. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Wu, Y.; Liu, T. Determination of the Redox Potentials of Solution and Solid Surface of Fe(II) Associated with Iron Oxyhydroxides. ACS Earth Sp. Chem. 2019, 3, 711–717. [Google Scholar] [CrossRef]
- Feng, Z.H.E.N.; Zhao, J.; Huggins, F.E.; Huffman, G.P. Agglomeration and phase transition of a nanophase iron oxide catalyst. J. Catal. 1993, 143, 510–519. [Google Scholar] [CrossRef]
- Lancee, R.J.; Dugulan, A.I.; Thüne, P.C.; Veringa, H.J.; Niemantsverdriet, J.W.; Fredriksson, H.O.A. Chemical looping capabilities of olivine, used as a catalyst in indirect biomass gasification. Appl. Catal. B Environ. 2014, 145, 216–222. [Google Scholar] [CrossRef]
- Tamaura, Y.; Ito, K.; Katsura, T. Y-FeO(OH) to Fe3O4. J. Chem. Soc. Dalt. Trans. 1983, 409, 189–194. [Google Scholar] [CrossRef]
- Spreitzer, D.; Schenk, J. Reduction of Iron Oxides with Hydrogen—A Review. Steel Res. Int. 2019, 90, 1900108. [Google Scholar] [CrossRef] [Green Version]
- Tamura, H. The role of rusts in corrosion and corrosion protection of iron and steel. Corros. Sci. 2008, 50, 1872–1883. [Google Scholar] [CrossRef] [Green Version]
- Baolin, H.; Zhang, H.; Hongzhong, L.I.; Qingshan, Z.H.U. Study on Kinetics of Iron Oxide Reduction by Hydrogen. Chin. J. Chem. Eng. 2012, 20, 10–17. [Google Scholar]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Mullet, M.; Khare, V.; Ruby, C. XPS study of Fe(II)-Fe(III) (oxy)hydroxycarbonate green rust compounds. Surf. Interface Anal. 2008, 40, 323–328. [Google Scholar] [CrossRef]
- Preisinger, M.; Krispin, M.; Rudolf, T.; Horn, S.; Strongin, D.R. Electronic structure of nanoscale iron oxide particles measured by scanning tunneling and photoelectron spectroscopies. Phys. Rev. B 2005, 71, 165409. [Google Scholar] [CrossRef] [Green Version]
- Wright, K.D.; Barron, A.R. Catalyst Residue and Oxygen Species Inhibition of the Formation of Hexahapto-Metal Complexes of Group 6 Metals on Single-Walled Carbon Nanotubes. J. Carbon Res. 2017, 3, 17. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Liu, Y.; Yang, L.; Tian, K.; He, L.; Zhang, Z.; Fang, S. Bimetallic metal–organic framework derived FeOx/TiO2 embedded in mesoporous carbon nanocomposite for the sensitive electrochemical detection of 4-nitrophenol. Sens. Actuators B Chem. 2019, 281, 1063–1072. [Google Scholar] [CrossRef]
- Fujii, T.; De Groot, F.M.F.; Sawatzky, G.A.; Voogt, F.C.; Hibma, T.; Okada, K. In Situ xps analysis of various iron oxide films grown by (formula presented)-assisted molecular-beam epitaxy. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 3195–3202. [Google Scholar] [CrossRef] [Green Version]
- Grosvenor, A.P.; Kobe, B.A.; Biesinger, M.C.; McIntyre, N.S. Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf. Interface Anal. 2004, 36, 1564–1574. [Google Scholar] [CrossRef]
- Yamashita, T.; Hayes, P. Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials. Appl. Surf. Sci. 2008, 254, 2441–2449. [Google Scholar] [CrossRef]
- Patwardhan, P.R.; Brown, R.C.; Shanks, B.H. Understanding the fast pyrolysis of lignin. ChemSusChem 2011, 4, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Wang, H.; Lin, H.; Zheng, Y. Selective production of guaiacol from black liquor: Effect of solvents. Carbon Resour. Convers. 2019, 2, 1–12. [Google Scholar] [CrossRef]
- Sajdak, M.; Chrubasik, M.; Muzyka, R. Chemical characterisation of tars from the thermal conversion of biomass by 1D and 2D gas chromatography combined with silylation. J. Anal. Appl. Pyrolysis 2017, 124, 426–438. [Google Scholar] [CrossRef]
- Kang, X.; Kirui, A.; Widanage, M.C.D.; Mentink-Vigier, F.; Cosgrove, D.J.; Wang, T. Lignin-polysaccharide interactions in plant secondary cell walls revealed by solid-state NMR. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Lupoi, J.S.; Singh, S.; Parthasarathi, R.; Simmons, B.A.; Henry, R.J. Recent innovations in analytical methods for the qualitative and quantitative assessment of lignin. Renew. Sustain. Energy Rev. 2015, 49, 871–906. [Google Scholar] [CrossRef] [Green Version]
- Laskar, D.D.; Yang, B.; Wang, H.; Lee, J. Pathways for biomass-derived lignin to hydrocarbon fuels. Biofuels Bioprod. Biorefin. 2013, 7, 602–626. [Google Scholar] [CrossRef]
- Guisnet, M.; Pinard, L. Characterization of acid-base catalysts through model reactions. Catal. Rev. Sci. Eng. 2018, 60, 337–436. [Google Scholar] [CrossRef]
- Kissin, Y.V. Chemical Mechanisms of Catalytic Cracking over Solid Acidic Catalysts: Alkanes and Alkenes. Catal. Rev. Sci. Eng. 2001, 43, 85–146. [Google Scholar] [CrossRef]
- De Lasa, H.I. Riser Simulator. U.S. Patent 5102628, 7 April 1992. [Google Scholar]
- Salaices, E. Catalytic Steam Gasification of Biomass Surrogates: A Thermodynamic and Kinetic Modelling. Ph.D. Thesis, The University of Western Ontario, London, ON, Canada, 2010. [Google Scholar]
- Mazumder, J. Steam Gasification of Biomass Surrogates: Catalyst. Ph.D. Thesis, The University of Western Ontario, London, ON, Canada, 2014. [Google Scholar]
- Quddus, M.R.; Hossain, M.M.; de Lasa, H.I. Ni based oxygen carrier over γ-Al2O3 for chemical looping combustion: Effect of preparation method on metal support interaction. Catal. Today 2013, 210, 124–134. [Google Scholar] [CrossRef]
- Mazumder, J.; de Lasa, H.I. Catalytic steam gasification of biomass surrogates: Thermodynamics and effect of operating conditions. Chem. Eng. J. 2016, 293, 232–242. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Support/Catalyst | SBET (m2/g) | VPore (cm3/g) | Avg. Pore Diameter (Å) |
|---|---|---|---|
| γ-Al2O3 | 193 | 0.51 | 139 |
| 5% CaO/ γ-Al2O3 | 143 | 0.45 | 131 |
| 10% CaO/ γ-Al2O3 | 118 | 0.36 | 126 |
| 4% Fe/10%CaO/ γ-Al2O3 | 120 | 0.39 | 131 |
| Support/ Catalyst Sample | cm3/g STP | μmol/m2 | μmol/g Solid |
|---|---|---|---|
| γ-Al2O3 | 11.31 | 2.62 | 505.06 |
| 5% CaO/ γ-Al2O3 | 6.40 | 2.00 | 285.55 |
| 10% CaO/ γ-Al2O3 | 4.25 | 1.61 | 189.94 |
| FexOy/10% CaO/ γ-Al2O3 | 5.06 | 1.88 | 225.94 |
| γ-Al2O3 | 5%CaO−γ-Al2O3 | 10%CaO−γ-Al2O3 | Catalyst | |||||
|---|---|---|---|---|---|---|---|---|
| Acidity Type | Area (cm3 STP NH3) | *MT (°C) | Area (cm3 STP NH3) | *MT (°C) | Area (cm3 STP NH3) | *MT (°C) | Area (cm3 STP NH3) | *MT (°C) |
| Weak | 5.54 | 183.7 | 4.36 | 177.3 | 2.96 | 177.8 | 3.74 | 178.1 |
| Medium | 2.36 | 246.2 | 2.05 | 245.9 | 1.31 | 223.2 | 0 | |
| Strong | 2.26 | 326.5 | 0 | 0 | 0.86 | 337.9 | ||
| Very Strong | 1.15 | 461.1 | 0 | 0 | 0.51 | 461.5 | ||
| Sample Condition | Temperature (°C) | Quantity (cm3/g STP) |
|---|---|---|
| Fresh Catalyst | 950 | 24.13 |
| Fresh Catalyst | 650 | 15.15 |
| Regenerated | 650 | 14.62 |
| Fresh 650 °C a | Regenerated 650 °C b | Fresh 950 °C c | ||||||
|---|---|---|---|---|---|---|---|---|
| R2 = 0.9996 | R2 = 0.9996 | R2 =0.9995 | ||||||
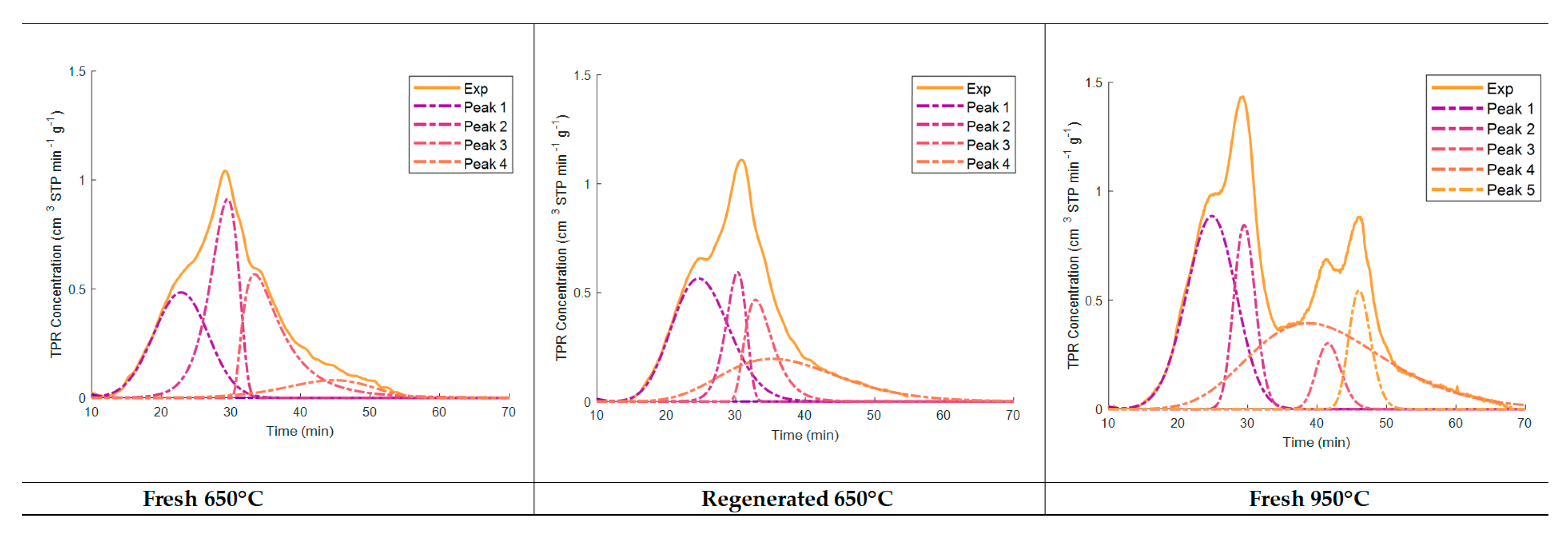
| Area (cm3 STP g−1) | Max. Peak (min) | Area (cm3 STP g−1) | Max. Peak (min) | Area (cm3 STP g−1) | Max. Peak (min) | |||
| Peak 1 | 4.73 | 22.8 | Peak 1 | 5.83 | 24.5 | Peak 1 | 7.89 | 24.8 |
| Peak 2 | 4.83 | 29.5 | Peak 2 | 2.21 | 30.2 | Peak 2 | 3.28 | 29.5 |
| Peak 3 | 4.33 | 33.3 | Peak 3 | 2.44 | 32.9 | Peak 3 | 1.32 | 38.3 |
| Peak 4 | 1.26 | 45.1 | Peak 4 | 4.14 | 35.7 | Peak 4 | 9.51 | 41.5 |
| Peak 5 | 2.14 | 46.0 | ||||||
| Total | 15.15 | 14.62 | 24.13 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, C.; Rostom, S.; de Lasa, H. An Eco-Friendly Fluidizable FexOy/CaO-γ-Al2O3 Catalyst for Tar Cracking during Biomass Gasification. Catalysts 2020, 10, 806. https://doi.org/10.3390/catal10070806
Torres C, Rostom S, de Lasa H. An Eco-Friendly Fluidizable FexOy/CaO-γ-Al2O3 Catalyst for Tar Cracking during Biomass Gasification. Catalysts. 2020; 10(7):806. https://doi.org/10.3390/catal10070806
Chicago/Turabian StyleTorres, Cindy, Samira Rostom, and Hugo de Lasa. 2020. "An Eco-Friendly Fluidizable FexOy/CaO-γ-Al2O3 Catalyst for Tar Cracking during Biomass Gasification" Catalysts 10, no. 7: 806. https://doi.org/10.3390/catal10070806
APA StyleTorres, C., Rostom, S., & de Lasa, H. (2020). An Eco-Friendly Fluidizable FexOy/CaO-γ-Al2O3 Catalyst for Tar Cracking during Biomass Gasification. Catalysts, 10(7), 806. https://doi.org/10.3390/catal10070806