

The C-H Bond Activation Triggered by Subsurface Mo Dopant on MgO Catalyst in Oxidative Coupling of Methane



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
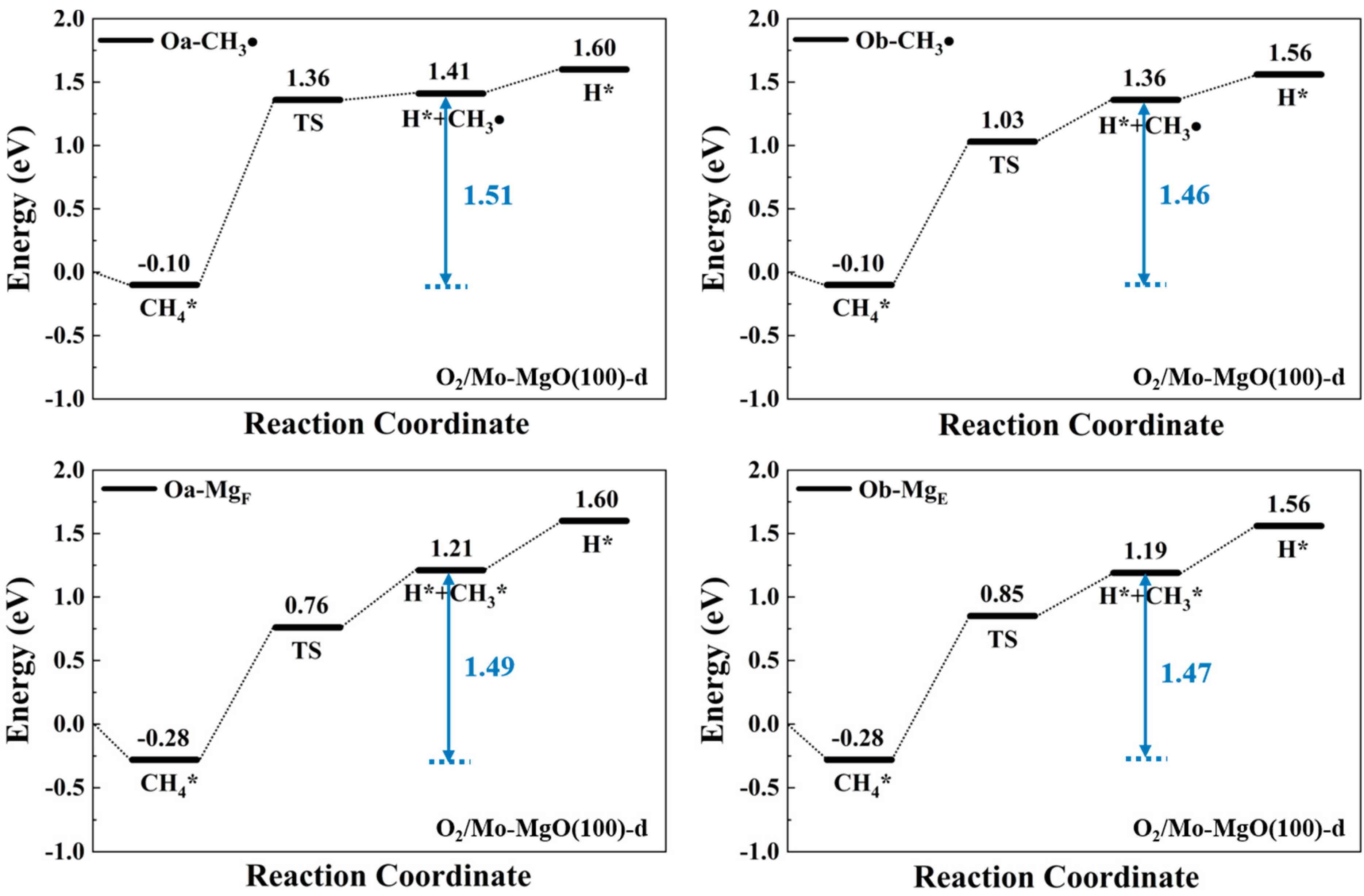
2. Results and Discussion
3. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Taifan, W.; Baltrusaitis, J. CH4 Conversion to Value Added Products: Potential, Limitations and Extensions of a Single Step Heterogeneous Catalysis. Appl. Catal. B Environ. 2016, 198, 525–547. [Google Scholar] [CrossRef]
- Meng, X.; Cui, X.; Rajan, N.P.; Yu, L.; Deng, D.; Bao, X. Direct Methane Conversion under Mild Condition by Thermo-, Electro-, or Photocatalysis. Chem 2019, 5, 2296–2325. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef] [PubMed]
- Rogge, T.; Kaplaneris, N.; Chatani, N.; Kim, J.; Chang, S.; Punji, B.; Schafer, L.L.; Musaev, D.G.; Wencel-Delord, J.; Roberts, C.A.; et al. C–H Activation. Nat. Rev. Methods Primers 2021, 1, 43. [Google Scholar] [CrossRef]
- Dalton, T.; Faber, T.; Glorius, F. C–H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 7, 245–261. [Google Scholar] [CrossRef]
- Panda, B. Joy and Challenges of Alkynylation of Arenes and Heteroarenes through Double C−H Functionalizations. Asian J. Org. Chem. 2020, 9, 492–507. [Google Scholar] [CrossRef]
- Hutchings, G.J.; Scurrell, M.S.; Woodhouse, J.R. Oxidative Coupling of Methane Using Oxide Catalysts. Chem. Soc. Rev. 1989, 18, 251–283. [Google Scholar] [CrossRef]
- Gambo, Y.; Jalil, A.A.; Triwahyono, S.; Abdulrasheed, A.A. Recent Advances and Future Prospect in Catalysts for Oxidative Coupling of Methane to Ethylene: A Review. J. Ind. Eng. Chem. 2018, 59, 218–229. [Google Scholar] [CrossRef]
- Postma, R.S.; Keijsper, D.J.; Morsink, B.F.; Siegers, E.H.; Mercimek, M.E.E.; Nieukoop, L.K.; van den Berg, H.; van der Ham, A.G.J.; Lefferts, L. Technoeconomic Evaluation of the Industrial Implementation of Catalytic Direct Nonoxidative Methane Coupling. Ind. Eng. Chem. Res. 2021, 61, 566–579. [Google Scholar] [CrossRef]
- Lunsford, J.H. The Catalytic Oxidative Coupling of Methane. Angew. Chem. Int. Ed. 1995, 34, 970–980. [Google Scholar] [CrossRef]
- Lin, C.H.; Ito, T.; Wang, J.; Lunsford, J.H. Oxidative Dimerization of Methane over Magnesium and Calcium Oxide Catalysts Promoted with Group IA Ions: The Role of [M+O−] Centers. J. Am. Chem. Soc. 1987, 109, 4808–4810. [Google Scholar] [CrossRef]
- Campbell, K.D.; Lunsford, J.H. Contribution of Gas-Phase Radical Coupling in the Catalytic Oxidation of Methane. J. Phys. Chem. 1988, 92, 5792–5796. [Google Scholar] [CrossRef]
- Kwapien, K.; Paier, J.; Sauer, J.; Geske, M.; Zavyalova, U.; Horn, R.; Schwach, P.; Trunschke, A.; Schlögl, R. Sites for Methane Activation on Lithium-Doped Magnesium Oxide Surfaces. Angew. Chem. Int. Ed. 2014, 53, 8774–8778. [Google Scholar] [CrossRef]
- Schwach, P.; Hamilton, N.; Eichelbaum, M.; Thum, L.; Lunkenbein, T.; Schlögl, R.; Trunschke, A. Structure Sensitivity of the Oxidative Activation of Methane over MgO Model Catalysts: II. Nature of Active Sites and Reaction Mechanism. J. Catal. 2015, 329, 574–587. [Google Scholar] [CrossRef]
- Zavyalova, U.; Weinberg, G.; Frandsen, W.; Girgsdies, F.; Risse, T.; Dinse, K.P.; Schloegl, R.; Horn, R. Lithium as a Modifier for Morphology and Defect Structure of Porous Magnesium Oxide Materials Prepared by Gel Combustion Synthesis. ChemCatChem 2011, 3, 1779–1788. [Google Scholar] [CrossRef]
- Cui, Y.; Shao, X.; Baldofski, M.; Sauer, J.; Nilius, N.; Freund, H.-J.J. Adsorption, Activation, and Dissociation of Oxygen on Doped Oxides. Angew. Chem. Int. Ed. 2013, 52, 11385–11387. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Uberuaga, B.P.; Jonsson, H. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Li, X.; Liu, Y.; Yu, Z.; Li, B.; Zhao, Z. The C-H Bond Activation Triggered by Subsurface Mo Dopant on MgO Catalyst in Oxidative Coupling of Methane. Catalysts 2022, 12, 1083. https://doi.org/10.3390/catal12101083
Sun X, Li X, Liu Y, Yu Z, Li B, Zhao Z. The C-H Bond Activation Triggered by Subsurface Mo Dopant on MgO Catalyst in Oxidative Coupling of Methane. Catalysts. 2022; 12(10):1083. https://doi.org/10.3390/catal12101083
Chicago/Turabian StyleSun, Xiaoying, Xinyu Li, Yue Liu, Zhan Yu, Bo Li, and Zhen Zhao. 2022. "The C-H Bond Activation Triggered by Subsurface Mo Dopant on MgO Catalyst in Oxidative Coupling of Methane" Catalysts 12, no. 10: 1083. https://doi.org/10.3390/catal12101083
APA StyleSun, X., Li, X., Liu, Y., Yu, Z., Li, B., & Zhao, Z. (2022). The C-H Bond Activation Triggered by Subsurface Mo Dopant on MgO Catalyst in Oxidative Coupling of Methane. Catalysts, 12(10), 1083. https://doi.org/10.3390/catal12101083