
Effect of Explicit Water Molecules on the Electrochemical Hydrogenation of CO2 on Sn(112)


Abstract
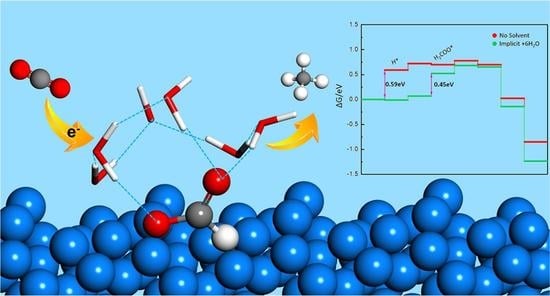
:
1. Introduction
2. Results and Discussion
2.1. Implicit Solvent Effect
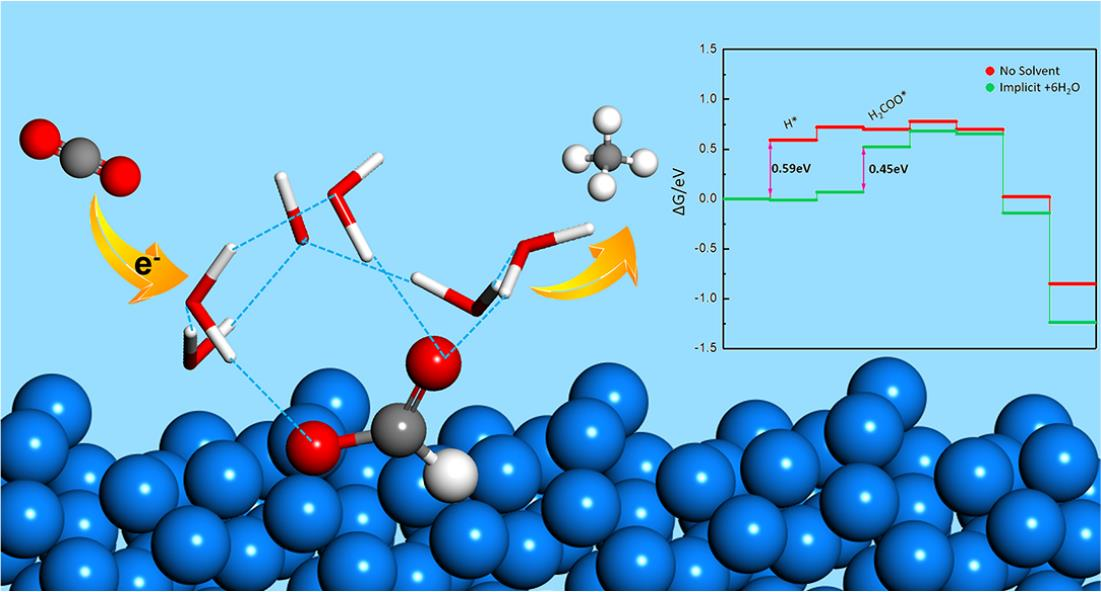
2.2. Explicit Solvent Effect
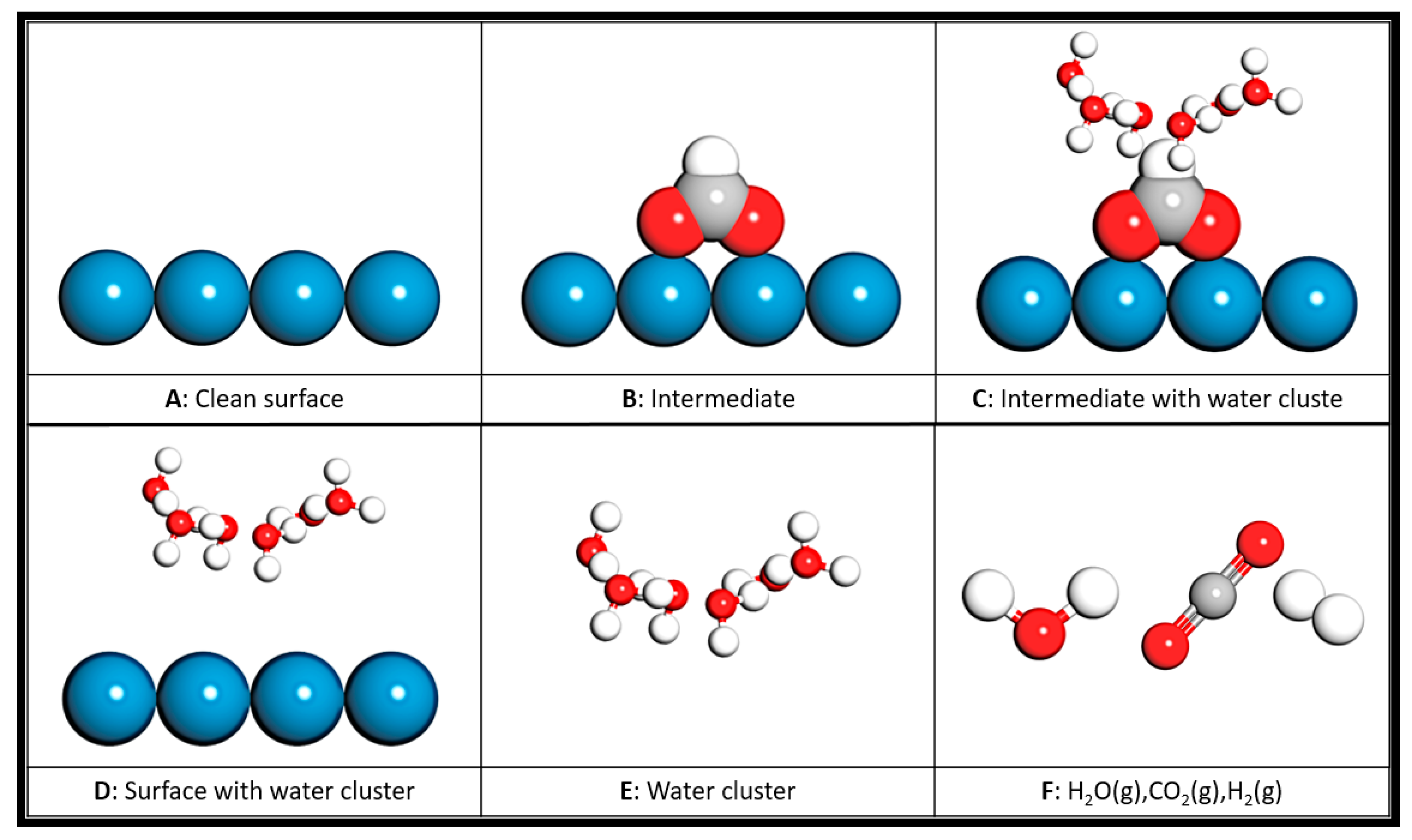
2.2.1. Interaction between Adsorbates and Explicit Water Molecules
2.2.2. Explicit Solvent Effect for the System
2.3. Combined Effect of Implicit and Explicit Solvents
2.4. Solvent Effect on the Potential Limiting Step and Limiting Potential
3. Methods
3.1. Density Functional Theory Calculations
3.2. Reference Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dattila, F.; Seemakurthi, R.R.; Zhou, Y.; López, N. Modeling Operando Electrochemical CO2 Reduction. Chem. Rev. 2022, 122, 11085–11130. [Google Scholar] [CrossRef]
- Wang, F.; Li, Y.; Xia, X.; Cai, W.; Chen, Q.; Chen, M. Metal–CO2 Electrochemistry: From CO2 Recycling to Energy Storage. Adv. Energy Mater. 2021, 11, 2100667. [Google Scholar] [CrossRef]
- Yang, X.; Zhuang, Y.; Zhu, J.; Le, J.; Cheng, J. Recent progress on multiscale modeling of electrochemistry. WIREs Comput. Mol. Sci. 2022, 12, e1559. [Google Scholar] [CrossRef]
- Sajid, A.; Pervaiz, E.; Ali, H.; Noor, T.; Baig, M.M. A perspective on development of fuel cell materials: Electrodes and electrolyte. Int. J. Energy Res. 2022, 46, 6953–6988. [Google Scholar] [CrossRef]
- Zhong, W.; Huang, W.; Ruan, S.; Zhang, Q.; Wang, Y.; Xie, S. Electrocatalytic Reduction of CO2 Coupled with Organic Conversion to Selectively Synthesize High-Value Chemicals. Chem. A Eur. J. 2022, 29, e202203228. [Google Scholar] [CrossRef]
- Al-Tamreh, S.A.; Ibrahim, M.H.; El-Naas, M.H.; Vaes, J.; Pant, D.; Benamor, A.; Amhamed, A. Electroreduction of Carbon Dioxide into Formate: A Comprehensive Review. ChemElectroChem 2021, 8, 3207–3220. [Google Scholar] [CrossRef]
- Xiao, C.; Zhang, J. Architectural Design for Enhanced C2 Product Selectivity in Electrochemical CO2 Reduction Using Cu-Based Catalysts: A Review. ACS Nano 2021, 15, 7975–8000. [Google Scholar] [CrossRef]
- Figueiredo, M.C.; Ledezma-Yanez, I.; Koper, M.T.M. In Situ Spectroscopic Study of CO2 Electroreduction at Copper Electrodes in Acetonitrile. ACS Catal. 2016, 6, 2382–2392. [Google Scholar] [CrossRef]
- Landaeta, E.; Kadosh, N.I.; Schultz, Z.D. Mechanistic Study of Plasmon-Assisted In Situ Photoelectrochemical CO2 Reduction to Acetate with a Ag/Cu2O Nanodendrite Electrode. ACS Catal. 2023, 13, 1638–1648. [Google Scholar] [CrossRef]
- Ibn Shamsah, S.M. Electrochemical Performance of Cupric Oxide Loaded Carbon Nanotubes as Electrode Material for CO2 Reduction. J. New Mater. Electrochem. Syst. 2021, 24, 9–13. [Google Scholar] [CrossRef]
- Li, M.; Li, W.; Song, W.; Wang, C.; Yao, Y.; Wu, C.; Luo, W.; Zou, Z. Do Cu Substrates Participate in Bi Electrocatalytic CO2 Reduction? ChemNanoMat 2021, 7, 128–133. [Google Scholar] [CrossRef]
- Silva, B.C.E.; Irikura, K.; Galvao Frem, R.C.; Boldrin Zanoni, M.V. Effect of Cu (BDC-NH2) MOF deposited on Cu/Cu2O electrode and its better performance in photoelectrocatalytic reduction of CO2. J. Electroanal. Chem. 2021, 880, 114856. [Google Scholar] [CrossRef]
- Chen, Y.; Wrubel, J.A.; Vise, A.E.; Intia, F.; Harshberger, S.; Klein, E.; Smith, W.A.; Ma, Z.; Deutsch, T.G.; Neyerlin, K.C. The effect of catholyte and catalyst layer binders on CO2 electroreduction selectivity. Chem. Catal. 2022, 2, 400–421. [Google Scholar] [CrossRef]
- Ao, C.; Feng, B.; Qian, S.; Wang, L.; Zhao, W.; Zhai, Y.; Zhang, L. Theoretical study of transition metals supported on g-C3N4 as electrochemical catalysts for CO2 reduction to CH3OH and CH4. J. CO2 Util. 2020, 36, 116–123. [Google Scholar] [CrossRef]
- Shi, N.; Yin, X.; Gao, W.; Wang, J.; Zhang, S.; Fan, Y.; Wang, M. Competition between electrocatalytic CO2 reduction and H+ reduction by Cu(II), Co(II) complexes containing redox-active ligand. Inorg. Chim. Acta 2021, 526, 120548. [Google Scholar] [CrossRef]
- Zhu, L.; Lin, Y.; Liu, K.; Cortes, E.; Li, H.; Hu, J.; Yamaguchi, A.; Liu, X.; Miyauchi, M.; Fu, J.; et al. Tuning the intermediate reaction barriers by a CuPd catalyst to improve the selectivity of CO2 electroreduction to C-2 products. Chin. J. Catal. 2021, 42, 1500–1508. [Google Scholar] [CrossRef]
- Zhang, X.G.; Zhao, Y.; Chen, S.; Xing, S.M.; Dong, J.C.; Li, J.F. Electrolyte effect for carbon dioxide reduction reaction on copper electrode interface: A DFT prediction. J. Chem. Phys. 2023, 158, 094704. [Google Scholar] [CrossRef]
- Zhai, L.; Cui, C.; Zhao, Y.; Zhu, X.; Han, J.; Wang, H.; Ge, Q. Titania-Modified Silver Electrocatalyst for Selective CO2 Reduction to CH3OH and CH4 from DFT Study. J. Phys. Chem. C 2017, 121, 16275–16282. [Google Scholar] [CrossRef]
- Sun, L.; Han, J.; Ge, Q.; Zhu, X.; Wang, H. Understanding the role of Cu+/Cu0 sites at Cu2O based catalysts in ethanol production from CO2 electroreduction -A DFT study. RSC Adv. 2022, 12, 19394–19401. [Google Scholar] [CrossRef]
- Masood, Z.; Ge, Q. Electrochemical reduction of CO2 at the earth-abundant transition metal-oxides/copper interfaces. Catal. Today 2023, 409, 53–62. [Google Scholar] [CrossRef]
- Varghese, J.J.; Mushrif, S.H. Origins of complex solvent effects on chemical reactivity and computational tools to investigate them: A review. React. Chem. Eng. 2019, 4, 165–206. [Google Scholar] [CrossRef]
- Du, Y.; An, W. Effects of Uniaxial Lattice Strain and Explicit Water Solvation on CO2 Electroreduction over a Cu Electrode: A Density Functional Theory Perspective. J. Phys. Chem. C 2021, 125, 9138–9149. [Google Scholar] [CrossRef]
- Sathishkumar, N.; Wu, S.; Chen, H. Mechanistic insights into chemical reduction of CO2 by reverse water-gas shift reaction on Ru(0001) surface: The water promotion effect. Appl. Surf. Sci. 2022, 581, 152354. [Google Scholar] [CrossRef]
- Wang, Y.X.; Zheng, M.; Wang, X.; Zhou, X. Electrocatalytic Reduction of CO2 to C-1 Compounds by Zn-Based Monatomic Alloys: A DFT Calculation. Catalysts 2022, 12, 1617. [Google Scholar] [CrossRef]
- Ludwig, T.; Gauthier, J.A.; Brown, K.S.; Ringe, S.; Norskov, J.K.; Chan, K. Solvent-Adsorbate Interactions and Adsorbate-Specific Solvent Structure in Carbon Dioxide Reduction on a Stepped Cu Surface. J. Phys. Chem. C 2019, 123, 5999–6009. [Google Scholar] [CrossRef] [Green Version]
- Garcia Ratés, M.; García Muelas, R.; López, N. Solvation Effects on Methanol Decomposition on Pd(111), Pt(111), and Ru(0001). J. Phys. Chem. C 2017, 121, 13803–13809. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, Z. Modeling the effect of an anion on the free energy surfaces along the reaction pathways of oxygen reduction on Pt(111). Chem. Phys. Lett. 2019, 736, 136813. [Google Scholar] [CrossRef]
- Briquet, L.G.V.; Sarwar, M.; Mugo, J.; Jones, G.; Calle-Vallejo, F. A New Type of Scaling Relations to Assess the Accuracy of Computational Predictions of Catalytic Activities Applied to the Oxygen Evolution Reaction. ChemCatChem 2017, 9, 1261–1268. [Google Scholar] [CrossRef]
- Sakong, S.; Groß, A. The Importance of the Electrochemical Environment in the Electro-Oxidation of Methanol on Pt(111). ACS Catal. 2016, 6, 5575–5586. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Nørskov, J.K.; Taylor, C.D.; Janik, M.J.; Neurock, M. Calculated Phase Diagrams for the Electrochemical Oxidation and Reduction of Water over Pt(111). J. Phys. Chem. B 2006, 110, 21833–21839. [Google Scholar] [CrossRef]
- Filhol, J.S.; Neurock, M. Elucidation of the Electrochemical Activation of Water over Pd by First Principles. Angew. Chem. Int. Ed. 2006, 45, 402–406. [Google Scholar] [CrossRef]
- Bu, Y.; Cui, T.; Zhao, M.; Zheng, W.; Gao, W.; Jiang, Q. Evolution of Water Structures on Stepped Platinum Surfaces. J. Phys. Chem. C 2018, 122, 604–611. [Google Scholar] [CrossRef]
- Mu, R.; Zhao, Z.; Dohnálek, Z.; Gong, J. Structural motifs of water on metal oxide surfaces. Chem. Soc. Rev. 2017, 46, 1785–1806. [Google Scholar] [CrossRef]
- Chen, J.; Schusteritsch, G.; Pickard, C.J.; Salzmann, C.G.; Michaelides, A. Two Dimensional Ice from First Principles: Structures and Phase Transitions. Phys. Rev. Lett. 2016, 116, 025501. [Google Scholar] [CrossRef] [Green Version]
- Björneholm, O.; Hansen, M.H.; Hodgson, A.; Liu, L.M.; Limmer, D.T.; Michaelides, A.; Pedevilla, P.; Rossmeisl, J.; Shen, H.; Tocci, G.; et al. Water at Interfaces. Chem. Rev. 2016, 116, 7698–7726. [Google Scholar] [CrossRef]
- Carrasco, J.; Hodgson, A.; Michaelides, A. A molecular perspective of water at metal interfaces. Nat. Mater. 2012, 11, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Kolb, M.J.; Wermink, J.; Calle Vallejo, F.; Juurlink, L.B.F.; Koper, M.T.M. Initial stages of water solvation of stepped platinum surfaces. Phys. Chem. Chem. Phys. 2016, 18, 3416–3422. [Google Scholar] [CrossRef] [Green Version]
- Farnesi Camellone, M.; Negreiros Ribeiro, F.; Szabova, L.; Tateyama, Y.; Fabris, S. Catalytic Proton Dynamics at the Water/Solid Interface of Ceria-Supported Pt Clusters. J. Am. Chem. Soc. 2016, 138, 11560–11567. [Google Scholar] [CrossRef]
- Heenen, H.H.; Gauthier, J.A.; Kristoffersen, H.H.; Ludwig, T.; Chan, K. Solvation at metal/water interfaces: An ab initio molecular dynamics benchmark of common computational approaches. J. Chem. Phys. 2020, 152, 144703. [Google Scholar] [CrossRef] [Green Version]
- Ringe, S.; Hörmann, N.G.; Oberhofer, H.; Reuter, K. Implicit Solvation Methods for Catalysis at Electrified Interfaces. Chem. Rev. 2022, 122, 10777–10820. [Google Scholar] [CrossRef]
- Bramley, G.A.; Nguyen, M.T.; Glezakou, V.A.; Rousseau, R.; Skylaris, C.K. Understanding Adsorption of Organics on Pt(111) in the Aqueous Phase: Insights from DFT Based Implicit Solvent and Statistical Thermodynamics Models. J. Chem. Theory Comput. 2022, 18, 1849–1861. [Google Scholar] [CrossRef]
- Iyemperumal, S.K.; Deskins, N.A. Evaluating Solvent Effects at the Aqueous/Pt(111) Interface. ChemPhysChem 2017, 18, 2171–2190. [Google Scholar] [CrossRef]
- Petrosyan, S.A.; Rigos, A.A.; Arias, T.A. Joint density-functional theory: Ab initio study of Cr2O3 surface chemistry in solution. J. Phys. Chem. B 2005, 109, 15436–15444. [Google Scholar] [CrossRef]
- Calle Vallejo, F.; de Morais, R.F.; Illas, F.; Loffreda, D.; Sautet, P. Affordable Estimation of Solvation Contributions to the Adsorption Energies of Oxygenates on Metal Nanoparticles. J. Phys. Chem. C 2019, 123, 5578–5582. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.X.; Bu, Y.F.; Gao, W.; Jiang, Q. CO2 Reduction Mechanism on the Pb(111) Surface: Effect of Solvent and Cations. J. Phys. Chem. C 2017, 121, 19767–19773. [Google Scholar] [CrossRef]
- Meng, Y.; Xu, Z.; Shen, Z.; Xia, Q.; Cao, Y.; Wang, Y.; Li, X. Understanding the water molecule effect in metal-free B-based electrocatalysts for electrochemical CO2 reduction. J. Mater. Chem. A 2022, 10, 6508–6522. [Google Scholar] [CrossRef]
- Valter, M.; Wickman, B.; Hellman, A. Solvent Effects for Methanol Electrooxidation on Gold. J. Phys. Chem. C 2021, 125, 1355–1360. [Google Scholar] [CrossRef]
- Cui, C.; Wang, H.; Zhu, X.; Han, J.; Ge, Q. A DFT study of CO2 electrochemical reduction on Pb(211) and Sn(112). Sci. China Chem. 2015, 58, 607–613. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Greeley, J.; Karlberg, G.S. Electrocatalysis and Catalyst Screening from Density Functional Theory Calculations. In Fuel Cell Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2009; Chapter 3; pp. 57–92. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Hansen, H.A.; Montoya, J.H.; Zhang, Y.-J.; Shi, C.; Peterson, A.A.; Nørskov, J.K. Electroreduction of Methanediol on Copper. Catal. Lett. 2013, 143, 631–635. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: London, UK, 1990. [Google Scholar]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, J.; Lian, L.; Zhang, G.; An, P.; Zeng, K.; He, H.; Yuan, T.; Huang, J.; Wang, L.; et al. Oxygen Vacancy-Reinforced Water-Assisted Proton Hopping for Enhanced Catalytic Hydrogenation. ACS Catal. 2023, 13, 2326–2334. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, X.; Wang, H.; Han, J.; Mei, D.; Ge, Q. Aqueous Phase Aldol Condensation of Formaldehyde and Acetone on Anatase TiO2(101) Surface: A Theoretical Investigation. ChemCatChem 2020, 12, 1220–1229. [Google Scholar] [CrossRef]
- Tayyebi, E.; Hussain, J.; Skúlason, E. Why do RuO2 electrodes catalyze electrochemical CO2 reduction to methanol rather than methane or perhaps neither of those? Chem. Sci. 2020, 11, 9542–9553. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, K.; Sundararaman, R.; Letchworth Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, K.; Kolluru, V.S.C.; Mula, S.; Steinmann, S.N.; Hennig, R.G. Implicit self-consistent electrolyte model in plane-wave density-functional theory. J. Chem. Phys. 2019, 151, 234101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Z.; Walker, B.; Michaelides, A. Quantum nature of the hydrogen bond. Proc. Natl. Acad. Sci. USA 2011, 108, 6369–6373. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.X.; Wang, G.C. A Systematic Theoretical Study of Water Gas Shift Reaction on Cu(111) and Cu(110): Potassium Effect. ACS Catal. 2019, 9, 2261–2274. [Google Scholar] [CrossRef]
- Kolb, M.J.; Farber, R.G.; Derouin, J.; Badan, C.; Calle-Vallejo, F.; Juurlink, L.B.F.; Killelea, D.R.; Koper, M.T.M. Double-Stranded Water on Stepped Platinum Surfaces. Phys. Rev. Lett. 2016, 116, 136101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psofogiannakis, G.; St-Amant, A.; Ternan, M. Methane oxidation mechanism on Pt(111): A cluster model DFT study. J. Phys. Chem. B 2006, 110, 24593–24605. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species |  | |||
|---|---|---|---|---|
| H1 | −0.32 | +0.10 | +0.06 | +0.13 |
| H2 | - | - | +0.02 | +0.59 |
| O1 | - | −1.17 | −1.09 | −1.08 |
| O2 | - | −1.17 | −1.12 | −1.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Cui, C.; Zhu, X.; Wang, H.; Ge, Q. Effect of Explicit Water Molecules on the Electrochemical Hydrogenation of CO2 on Sn(112). Catalysts 2023, 13, 1033. https://doi.org/10.3390/catal13071033
Wang J, Cui C, Zhu X, Wang H, Ge Q. Effect of Explicit Water Molecules on the Electrochemical Hydrogenation of CO2 on Sn(112). Catalysts. 2023; 13(7):1033. https://doi.org/10.3390/catal13071033
Chicago/Turabian StyleWang, Jia, Chaonan Cui, Xinli Zhu, Hua Wang, and Qingfeng Ge. 2023. "Effect of Explicit Water Molecules on the Electrochemical Hydrogenation of CO2 on Sn(112)" Catalysts 13, no. 7: 1033. https://doi.org/10.3390/catal13071033
APA StyleWang, J., Cui, C., Zhu, X., Wang, H., & Ge, Q. (2023). Effect of Explicit Water Molecules on the Electrochemical Hydrogenation of CO2 on Sn(112). Catalysts, 13(7), 1033. https://doi.org/10.3390/catal13071033