
Affinity and Structural Analysis of the U1A RNA Recognition Motif with Engineered Methionines to Improve Experimental Phasing

 ,
,
Abstract
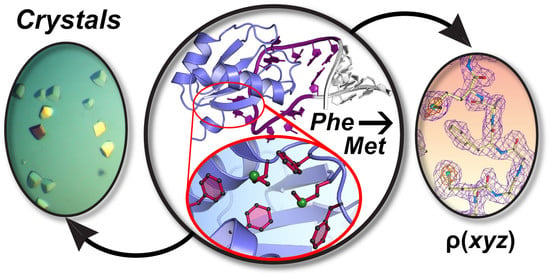
:
1. Introduction
2. Experimental Procedures
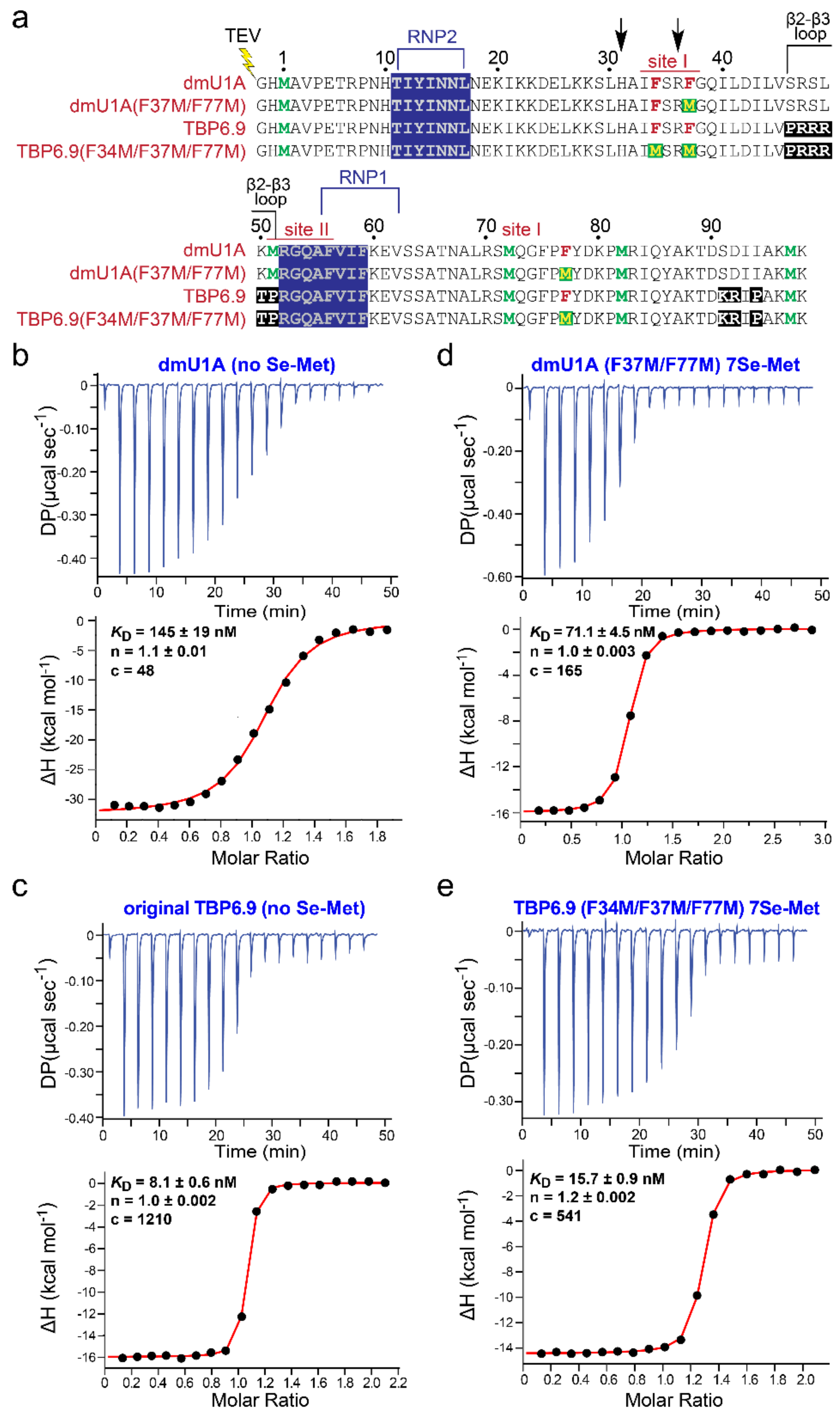
2.1. Expression of Se-Met-Labeled dmU1A(F37M/F77M) and TBP6.9(F34M/F37M/F77M)
2.2. Purification of dmU1A, TBP6.9, dmU1A(F37M/F77M), and TBP6.9(F34M/F37M/F77M)
2.3. ITC Analysis of dmU1A, dmU1A(F37M/F77M), TBP6.9, and TBP6.9 (F34M/F37M/F77M)
2.4. Crystallization, X-ray Diffraction Analysis, and Structure Determination of dmU1A(F37M/F77M)
2.5. Co-crystallization of Se-Met Labelled dmU1A(F37M/F77M) with Hpii and Riboswitch RNA
2.6. X-ray Diffraction Analysis of dmU1A(F37M/F77M) in Complex with a preQ1-II Riboswitch
3. Results and Discussion
3.1. Rationale of Phenylalanine-to-Methionine Mutants in the dmU1A Hydrophobic Core
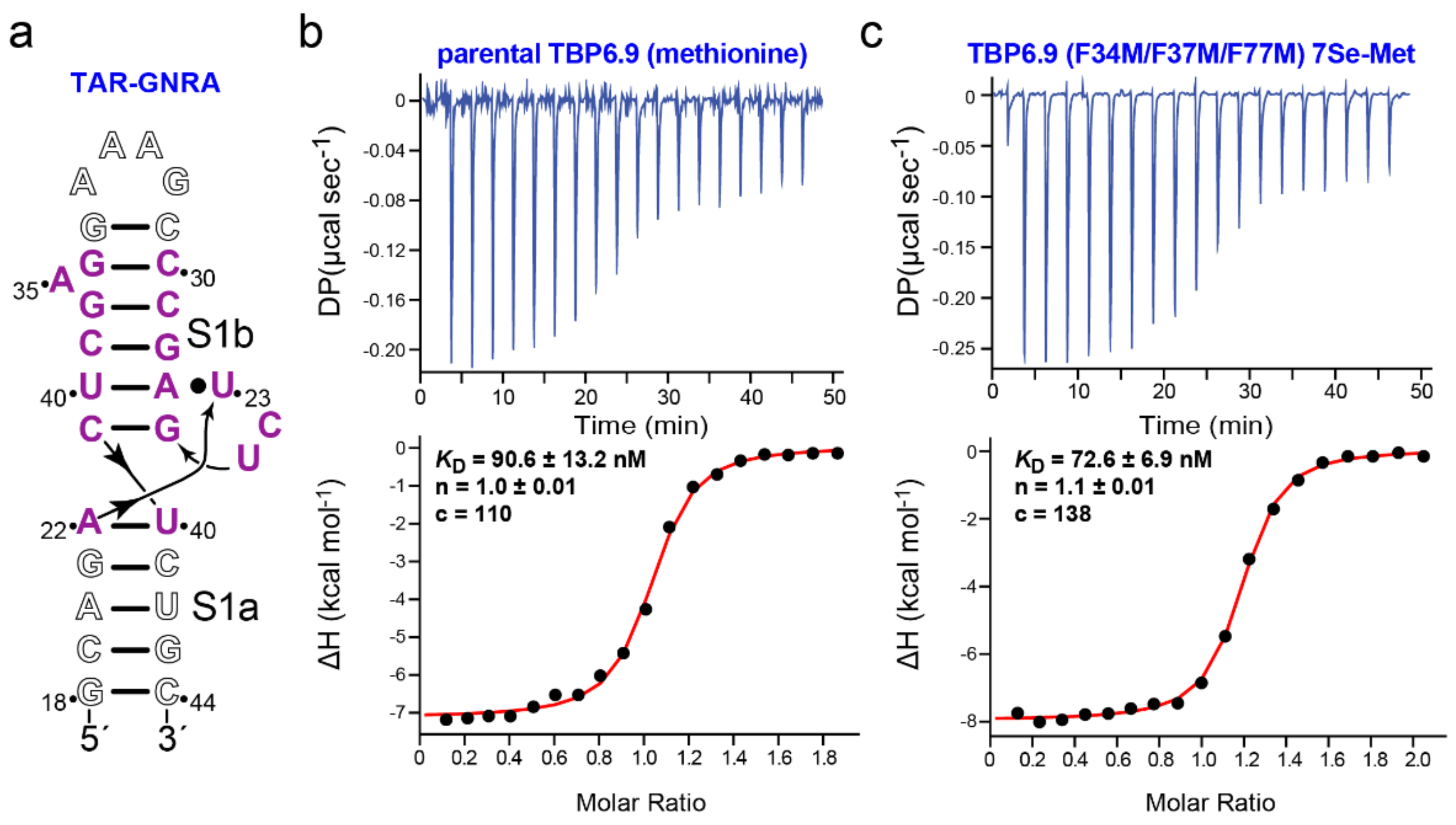
3.2. dmU1A(F37M/F77M) and TBP6.9(F34M/F37M/F77M) Retain Affinity for hpII and TAR RNAs
3.3. Evidence That TBP6.9(F34M/F37M/F77M) Targets a TAR Sequence Without the Apical Loop
3.4. Crystallization of dmU1A(F37M/F77M) Alone and in Complex with RNA
3.5. Structure Determination and Quality-Control Analysis of the dmU1A(F37M/F77M) Variant
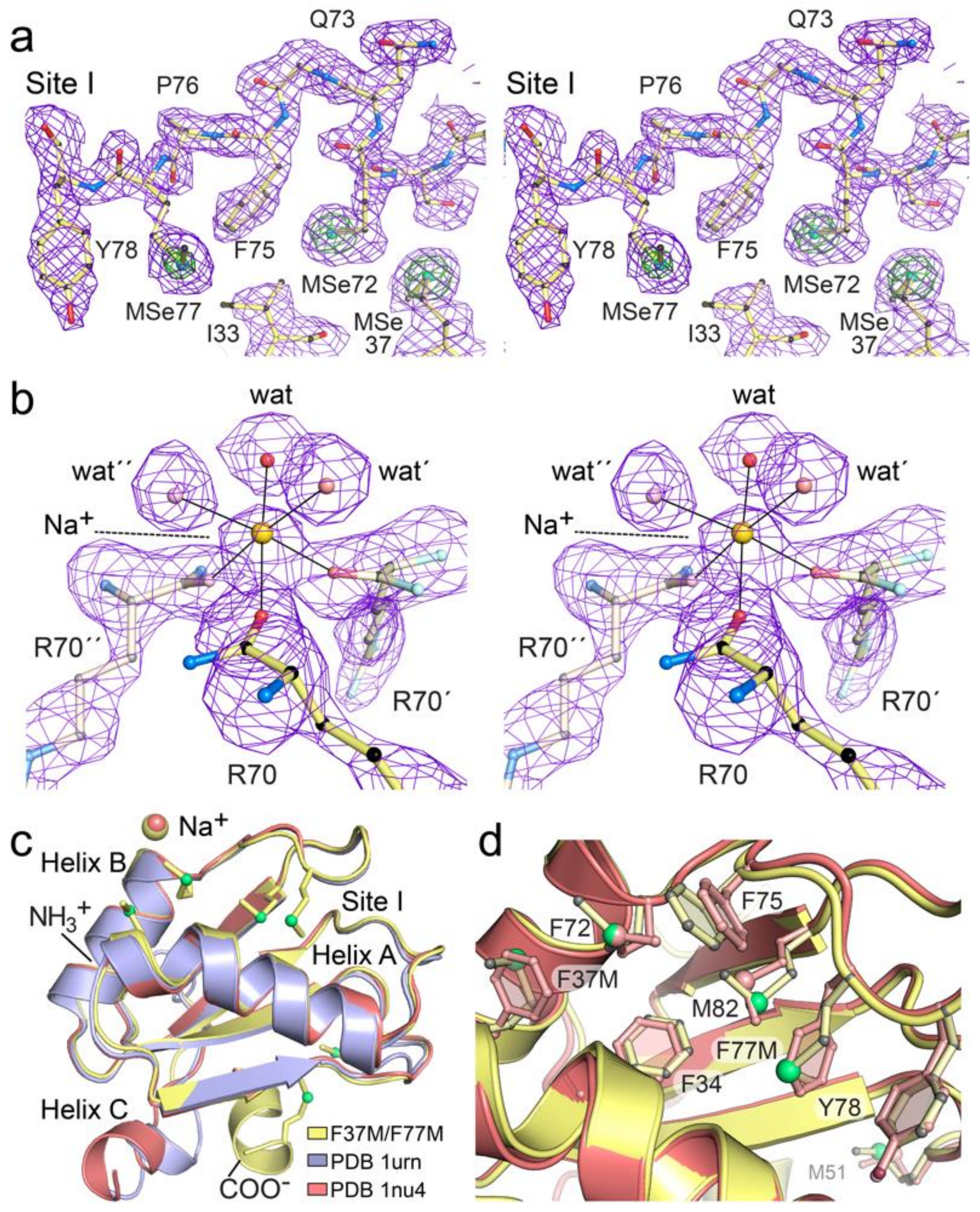
3.6. Comparison of the dmU1A(F37M/F77M) Variant to a dmU1A Structure Prepared from Malonate
3.7. Site I of dmU1A(F37M/F77M) Maintains the Core Fold but Makes New S–π Interactions
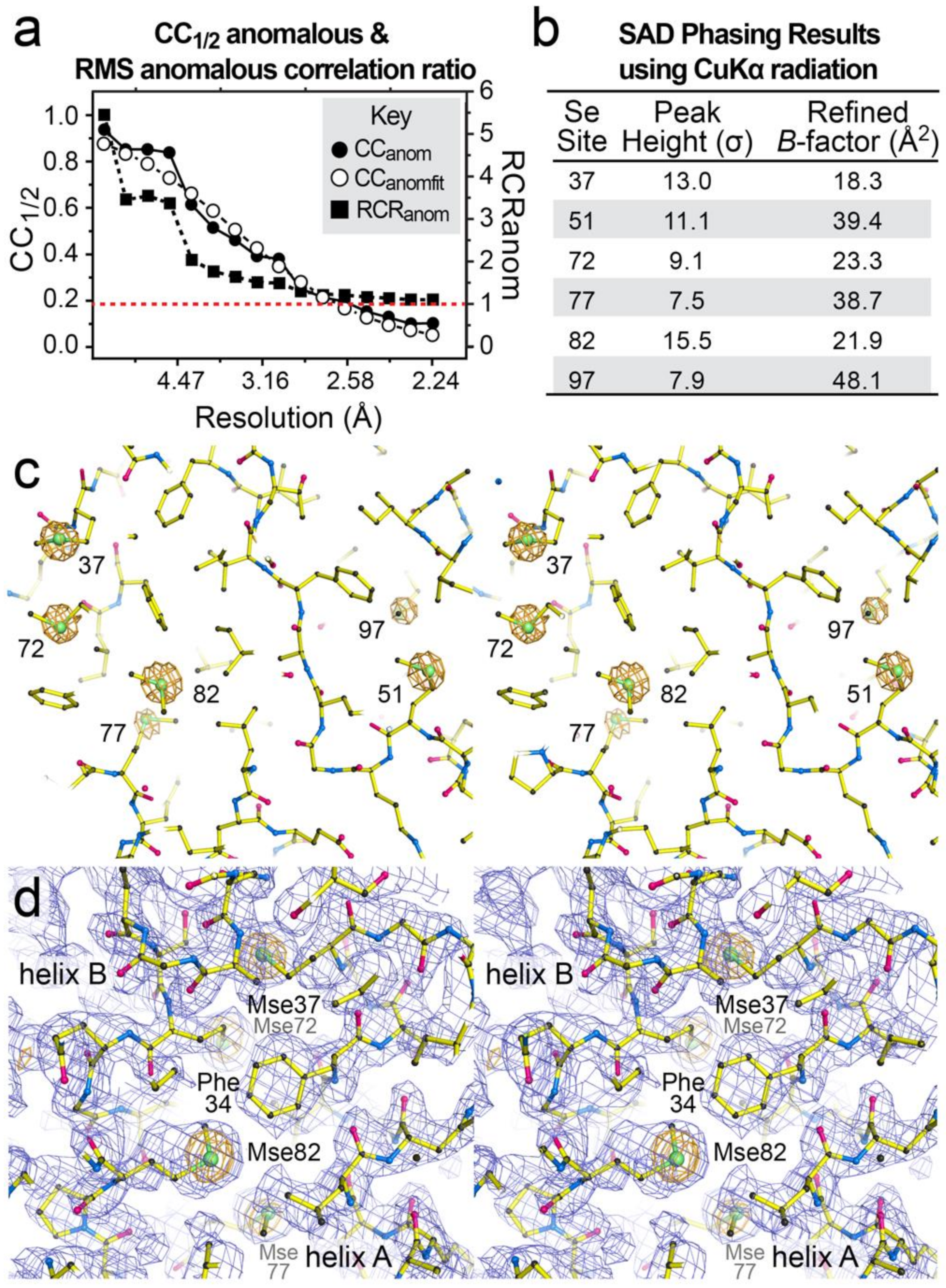
3.8. Substructure Determination and SAD Phasing of the dmU1A(F37M/F77M) Variant
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution—trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [Green Version]
- Pennisi, E. Encode project writes eulogy for junk DNA. Science 2012, 337, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, A.C.; Frank, A.T.; Kratz, J.D.; Swanson, M.D.; Gonzalez-Hernandez, M.J.; Lee, J.; Andricioaei, I.; Markovitz, D.M.; Al-Hashimi, H.M. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 2011, 7, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Disney, M.D.; Dwyer, B.G.; Childs-Disney, J.L. Drugging the RNA world. Cold Spring Harb. Perspect. Biol. 2018, 10, a034769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCown, P.J.; Corbino, K.A.; Stav, S.; Sherlock, M.E.; Breaker, R.R. Riboswitch diversity and distribution. RNA 2017, 23, 995–1011. [Google Scholar] [CrossRef]
- Shi, H.; Moore, P.B. The crystal structure of yeast phenylalanine tRNA at 1.93 Å resolution: A classic structure revisited. RNA 2000, 6, 1091–1105. [Google Scholar] [CrossRef] [Green Version]
- Fourmy, D.; Recht, M.I.; Blanchard, S.C.; Puglisi, J.D. Structure of the a site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science 1996, 274, 1367–1371. [Google Scholar] [CrossRef]
- Pley, H.W.; Flaherty, K.M.; McKay, D.B. Three-dimensional structure of a hammerhead ribozyme. Nature 1994, 372, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Wedekind, J.E.; McKay, D.B. Crystal structure of a lead-dependent ribozyme revealing metal binding sites relevant to catalysis. Nat. Struct. Biol. 1999, 6, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Theimer, C.A.; Blois, C.A.; Feigon, J. Structure of the human telomerase RNA pseudoknot reveals conserved tertiary interactions essential for function. Mol. Cell. 2005, 17, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Bulkley, D.; Wang, J.; Valenstein, M.L.; Yario, T.A.; Steitz, T.A.; Steitz, J.A. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat. Struct. Mol. Biol. 2014, 21, 633–640. [Google Scholar] [CrossRef]
- Toor, N.; Keating, K.S.; Taylor, S.D.; Pyle, A.M. Crystal structure of a self-spliced group II intron. Science 2008, 320, 77–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cate, J.H.; Gooding, A.R.; Podell, E.; Zhou, K.; Golden, B.L.; Kundrot, C.E.; Cech, T.R.; Doudna, J.A. Crystal structure of a group I ribozyme domain: Principles of RNA packing. Science 1996, 273, 1678–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correll, C.C.; Freeborn, B.; Moore, P.B.; Steitz, T.A. Metals, motifs, and recognition in the crystal structure of a 5S rRNA domain. Cell 1997, 91, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Correll, C.C.; Munishkin, A.; Chan, Y.L.; Ren, Z.; Wool, I.G.; Steitz, T.A. Crystal structure of the ribosomal RNA domain essential for binding elongation factors. Proc. Natl. Acad. Sci. USA 1998, 95, 13436–13441. [Google Scholar] [CrossRef] [Green Version]
- Golden, B.L.; Kim, H.; Chase, E. Crystal structure of a phage twort group I ribozyme-product complex. Nat. Struct. Mol. Biol. 2005, 12, 82–89. [Google Scholar] [CrossRef]
- Krasilnikov, A.S.; Yang, X.; Pan, T.; Mondragon, A. Crystal structure of the specificity domain of ribonuclease P. Nature 2003, 421, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Torres-Larios, A.; Swinger, K.K.; Krasilnikov, A.S.; Pan, T.; Mondragon, A. Crystal structure of the RNA component of bacterial ribonuclease P. Nature 2005, 437, 584–587. [Google Scholar] [CrossRef]
- Krasilnikov, A.S.; Mondragon, A. On the occurrence of the T-loop RNA folding motif in large RNA molecules. RNA 2003, 9, 640–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissen, P.; Ippolito, J.A.; Ban, N.; Moore, P.B.; Steitz, T.A. RNA tertiary interactions in the large ribosomal subunit: The A-minor motif. Proc. Natl. Acad. Sci. USA 2001, 98, 4899–4903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrix, D.K.; Brenner, S.E.; Holbrook, S.R. RNA structural motifs: Building blocks of a modular biomolecule. Q. Rev. Biophys. 2005, 38, 221–243. [Google Scholar] [CrossRef] [Green Version]
- Lilley, D.M. The k-turn motif in riboswitches and other RNA species. Biochim. Biophys. Acta 2014, 1839, 995–1004. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.A. Unraveling the structure and biological functions of RNA triple helices. Wiley Interdiscip. Rev. RNA 2020, 11, e1598. [Google Scholar] [CrossRef] [PubMed]
- Cate, J.H.; Gooding, A.R.; Podell, E.; Zhou, K.; Golden, B.L.; Szewczak, A.A.; Kundrot, C.E.; Cech, T.R.; Doudna, J.A. RNA tertiary structure mediation by adenosine platforms. Science 1996, 273, 1696–1699. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.; Peterson, R.; Feigon, J. Structural insights into riboswitch control of the biosynthesis of queuosine, a modified nucleotide found in the anticodon of tRNA. Mol. Cell. 2009, 33, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, G.M.; Dutta, D.; Cavender, C.E.; Jenkins, J.L.; Pritchett, E.M.; Baker, C.D.; Ashton, J.M.; Mathews, D.H.; Wedekind, J.E. Analysis of a preQ1-I riboswitch in effector-free and bound states reveals a metabolite-programmed nucleobase-stacking spine that controls gene regulation. Nucleic Acids Res. 2020, 48, 8146–8164. [Google Scholar] [CrossRef] [PubMed]
- Pley, H.W.; Flaherty, K.M.; McKay, D.B. Model for an RNA tertiary interaction from the structure of an intermolecular complex between a GAAA tetraloop and an RNA helix. Nature 1994, 372, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Ennifar, E.; Walter, P.; Ehresmann, B.; Ehresmann, C.; Dumas, P. Crystal structures of coaxially stacked kissing complexes of the HIV-1 RNA dimerization initiation site. Nat. Struct. Biol. 2001, 8, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Grum-Tokars, V.; Krucinska, J.; Kundracik, M.L.; Wedekind, J.E. Conformational heterogeneity at position U37 of an all-RNA hairpin ribozyme with implications for metal binding and the catalytic structure of the S-turn. Biochemistry 2005, 44, 14396–14408. [Google Scholar] [CrossRef]
- Chavali, S.S.; Bonn-Breach, R.; Wedekind, J.E. Face-time with TAR: Portraits of an HIV-1 RNA with diverse modes of effector recognition relevant for drug discovery. J. Biol. Chem. 2019, 294, 9326–9341. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Huang, H.; Qin, D.; Li, N.S.; Koide, A.; Staley, J.P.; Koide, S.; Kossiakoff, A.A.; Piccirilli, J.A. Specific recognition of a single-stranded RNA sequence by a synthetic antibody fragment. J. Mol. Biol. 2016, 428, 4100–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, A.; Gasser, C.; Li, F.; Chen, H.; Mair, S.; Krasheninina, O.; Micura, R.; Ren, A. Sam-VI riboswitch structure and signature for ligand discrimination. Nat. Commun. 2019, 10, 5728. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Egger, M.; Xu, X.; Flemmich, L.; Krasheninina, O.; Sun, A.; Micura, R.; Ren, A. Structural distinctions between NAD+ riboswitch domains 1 and 2 determine differential folding and ligand binding. Nucleic Acids Res. 2020, 48, 12394–12406. [Google Scholar] [CrossRef]
- Ke, A.; Zhou, K.; Ding, F.; Cate, J.H.; Doudna, J.A. A conformational switch controls hepatitis delta virus ribozyme catalysis. Nature 2004, 429, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Ferre-D’Amare, A.R.; Zhou, K.; Doudna, J.A. Crystal structure of a hepatitis delta virus ribozyme. Nature 1998, 395, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.L.; Stahley, M.R.; Kosek, A.B.; Wang, J.; Strobel, S.A. Crystal structure of a self-splicing group I intron with both exons. Nature 2004, 430, 45–50. [Google Scholar] [CrossRef]
- Ren, A.; Xue, Y.; Peselis, A.; Serganov, A.; Al-Hashimi, H.M.; Patel, D.J. Structural and dynamic basis for low-affinity, high-selectivity binding of L-glutamine by the glutamine riboswitch. Cell Rep. 2015, 13, 1800–1813. [Google Scholar] [CrossRef] [Green Version]
- Butler, E.B.; Xiong, Y.; Wang, J.; Strobel, S.A. Structural basis of cooperative ligand binding by the glycine riboswitch. Chem. Biol. 2011, 18, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.P.; Ferre-D’Amare, A.R. Crystal structure of a c-di-AMP riboswitch reveals an internally pseudo-dimeric RNA. EMBO J. 2014, 33, 2692–2703. [Google Scholar] [CrossRef]
- Kulshina, N.; Baird, N.J.; Ferre-D’Amare, A.R. Recognition of the bacterial second messenger cyclic diguanylate by its cognate riboswitch. Nat. Struct. Mol. Biol. 2009, 16, 1212–1217. [Google Scholar] [CrossRef]
- Smith, K.D.; Lipchock, S.V.; Ames, T.D.; Wang, J.; Breaker, R.R.; Strobel, S.A. Structural basis of ligand binding by a c-di-GMP riboswitch. Nat. Struct. Mol. Biol. 2009, 16, 1218–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Murakami, H.; Suga, H.; Ferre-D’Amare, A.R. Structural basis of specific tRNA aminoacylation by a small in vitro selected ribozyme. Nature 2008, 454, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Edwards, T.E.; Ferre-D’Amare, A.R. Structural basis for specific, high-affinity tetracycline binding by an in vitro evolved aptamer and artificial riboswitch. Chem. Biol. 2008, 15, 1125–1137. [Google Scholar] [CrossRef] [Green Version]
- Shechner, D.M.; Grant, R.A.; Bagby, S.C.; Koldobskaya, Y.; Piccirilli, J.A.; Bartel, D.P. Crystal structure of the catalytic core of an RNA-polymerase ribozyme. Science 2009, 326, 1271–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochrane, J.C.; Lipchock, S.V.; Smith, K.D.; Strobel, S.A. Structural and chemical basis for glucosamine 6-phosphate binding and activation of the glmS ribozyme. Biochemistry 2009, 48, 3239–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Lilley, D.M. Structure of a rare non-standard sequence k-turn bound by L7ae protein. Nucleic Acids Res. 2014, 42, 4734–4740. [Google Scholar] [CrossRef] [PubMed]
- Belashov, I.A.; Crawford, D.W.; Cavender, C.E.; Dai, P.; Beardslee, P.C.; Mathews, D.H.; Pentelute, B.L.; McNaughton, B.R.; Wedekind, J.E. Structure of HIV TAR in complex with a lab-evolved RRM provides insight into duplex RNA recognition and synthesis of a constrained peptide that impairs transcription. Nucleic Acids Res. 2018, 46, 6401–6415. [Google Scholar] [CrossRef]
- Chavali, S.S.; Mali, S.M.; Jenkins, J.L.; Fasan, R.; Wedekind, J.E. Co-crystal structures of HIV TAR RNA bound to lab-evolved proteins show key roles for arginine relevant to the design of cyclic peptide TAR inhibitors. J. Biol. Chem. 2020, 295, 16470–16486. [Google Scholar] [CrossRef] [PubMed]
- Ferre-D’Amare, A.R. Use of the spliceosomal protein U1A to facilitate crystallization and structure determination of complex RNAs. Methods 2010, 52, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferre-D’Amare, A.R.; Doudna, J.A. Crystallization and structure determination of a hepatitis delta virus ribozyme: Use of the RNA-binding protein U1A as a crystallization module. J. Mol. Biol. 2000, 295, 541–556. [Google Scholar] [CrossRef] [Green Version]
- Ferre-D’Amare, A.R. Use of the U1A protein to facilitate crystallization and structure determination of large RNAs. Methods Mol. Biol. 2016, 1320, 67–76. [Google Scholar]
- Oubridge, C.; Ito, N.; Evans, P.R.; Teo, C.H.; Nagai, K. Crystal structure at 1.92 Å resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature 1994, 372, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Oubridge, C.; Ito, N.; Teo, C.H.; Fearnley, I.; Nagai, K. Crystallisation of RNA-protein complexes. II. The application of protein engineering for crystallisation of the U1A protein-RNA complex. J. Mol. Biol. 1995, 249, 409–423. [Google Scholar] [CrossRef]
- Hendrickson, W.A. Anomalous diffraction in crystallographic phase evaluation. Q. Rev. Biophys. 2014, 47, 49–93. [Google Scholar] [CrossRef] [Green Version]
- Chavali, S.S.; Cavender, C.E.; Mathews, D.H.; Wedekind, J.E. Arginine forks are a widespread motif to recognize phosphate backbones and guanine nucleobases in the RNA major groove. J. Am. Chem. Soc. 2020, 142, 19835–19839. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, W. Maturation of MAD phasing for the determination of macromolecular structures. J. Synchroton Radiat. 1999, 6, 845–851. [Google Scholar] [CrossRef]
- Hendrickson, W.A.; Ogata, C.M. [28] Phase determination from multiwavelength anomalous diffraction measurements. Methods Enzymol. 1997, 276, 494–523. [Google Scholar] [PubMed]
- Smith, J.L. Multiwavelength anomalous diffraction in macromolecular crystallography. In Direct Methods for Solving Macromolecular Structures NATO ASI Series; Fortier, S., Ed.; Series C: Mathematical and Physical Sciences; Springer: Dordrecht, The Netherlands, 1998; pp. 211–225. [Google Scholar]
- Liberman, J.A.; Salim, M.; Krucinska, J.; Wedekind, J.E. Structure of a class II preQ1 riboswitch reveals ligand recognition by a new fold. Nat. Chem. Biol. 2013, 9, 353–355. [Google Scholar] [CrossRef] [Green Version]
- Doublie, S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997, 276, 523–530. [Google Scholar]
- Kapust, R.B.; Tozser, J.; Fox, J.D.; Anderson, D.E.; Cherry, S.; Copeland, T.D.; Waugh, D.S. Tobacco etch virus protease: Mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001, 14, 993–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippa, G.M.; Liberman, J.A.; Jenkins, J.L.; Krucinska, J.; Salim, M.; Wedekind, J.E. Crystallographic analysis of small ribozymes and riboswitches. Methods Mol. Biol. 2012, 848, 159–184. [Google Scholar]
- Liberman, J.A.; Bogue, J.T.; Jenkins, J.L.; Salim, M.; Wedekind, J.E. ITC analysis of ligand binding to preQ(1) riboswitches. Methods Enzymol. 2014, 549, 435–450. [Google Scholar] [PubMed] [Green Version]
- Fernando, H.; Chin, C.; Rosgen, J.; Rajarathnam, K. Dimer dissociation is essential for Interleukin-8 (Il-8) binding to CXCR1 receptor. J. Biol Chem. 2004, 279, 36175–36178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiseman, T.; Williston, S.; Brandts, J.F.; Lin, L.N. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 1989, 179, 131–137. [Google Scholar] [CrossRef]
- Turnbull, W.B.; Daranas, A.H. On the value of c: Can low affinity systems be studied by isothermal titration calorimetry? J. Am. Chem. Soc. 2003, 125, 14859–14866. [Google Scholar] [CrossRef]
- Benvenuti, M.; Mangani, S. Crystallization of soluble proteins in vapor diffusion for x-ray crystallography. Nat. Protoc. 2007, 2, 1633–1651. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with Phenix.Refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Grosse-Kunstleve, R.W.; Adams, P.D. Substructure search procedures for macromolecular structures. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 1966–1973. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T. Solve and Resolve: Automated structure solution, density modification and model building. J. Synchrotron Radiat. 2004, 11, 49–52. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. Sect. A 1978, 32, 922–923. [Google Scholar] [CrossRef]
- McPherson, A. Introduction to protein crystallization. Methods 2004, 34, 254–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitale, R.C.; Torelli, A.T.; Krucinska, J.; Bandarian, V.; Wedekind, J.E. The structural basis for recognition of the preQ0 metabolite by an unusually small riboswitch aptamer domain. J. Biol. Chem. 2009, 284, 11012–11016. [Google Scholar] [CrossRef] [Green Version]
- Otwinowski, Z.; Minor, W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Gassner, N.C.; Baase, W.A.; Hausrath, A.C.; Matthews, B.W. Substitution with selenomethionine can enhance the stability of methionine-rich proteins. J. Mol. Biol. 1999, 294, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Valley, C.C.; Cembran, A.; Perlmutter, J.D.; Lewis, A.K.; Labello, N.P.; Gao, J.; Sachs, J.N. The methionine-aromatic motif plays a unique role in stabilizing protein structure. J. Biol. Chem. 2012, 287, 34979–34991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, K.S.C.; Lindley, P.F.; Thornton, J.M. Sulphur-aromatic interactions in proteins. FEBS Lett. 1985, 190, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Katsamba, P.S.; Bayramyan, M.; Haworth, I.S.; Myszka, D.G.; Laird-Offringa, I.A. Complex role of the beta 2-beta 3 loop in the interaction of U1A with U1 hairpin II RNA. J. Biol. Chem. 2002, 277, 33267–33274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, D.W.; Blakeley, B.D.; Chen, P.H.; Sherpa, C.; Le Grice, S.F.; Laird-Offringa, I.A.; McNaughton, B.R. An evolved RNA recognition motif that suppresses HIV-1 Tat/TAR-dependent transcription. ACS Chem. Biol. 2016, 11, 2206–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberman, J.A.; Suddala, K.C.; Aytenfisu, A.; Chan, D.; Belashov, I.A.; Salim, M.; Mathews, D.H.; Spitale, R.C.; Walter, N.G.; Wedekind, J.E. Structural analysis of a class III preQ1 riboswitch reveals an aptamer distant from a ribosome-binding site regulated by fast dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, E3485–E3494. [Google Scholar] [CrossRef] [Green Version]
- Blose, J.M.; Proctor, D.J.; Veeraraghavan, N.; Misra, V.K.; Bevilacqua, P.C. Contribution of the closing base pair to exceptional stability in RNA tetraloops: Roles for molecular mimicry and electrostatic factors. J. Am. Chem. Soc. 2009, 131, 8474–8484. [Google Scholar] [CrossRef] [PubMed]
- Rupert, P.B.; Xiao, H.; Ferre-D’Amare, A.R. U1A RNA-binding domain at 1.8 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 1521–1524. [Google Scholar] [CrossRef] [PubMed]
- Rosenbach, H.; Victor, J.; Borggrafe, J.; Biehl, R.; Steger, G.; Etzkorn, M.; Span, I. Expanding crystallization tools for nucleic acid complexes using U1A protein variants. J. Struct. Biol. 2020, 210, 107480. [Google Scholar] [CrossRef] [PubMed]
- Wedekind, J.E.; McKay, D.B. Purification, crystallization, and x-ray diffraction analysis of small ribozymes. Methods Enzymol. 2000, 317, 149–168. [Google Scholar] [PubMed]
- McPherson, A. A comparison of salts for the crystallization of macromolecules. Protein Sci. 2001, 10, 418–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichenberger, C.X.; Rupp, B. Ten years of probabilistic estimates of biocrystal solvent content: New insights via nonparametric kernel density estimate. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1579–1588. [Google Scholar] [CrossRef]
- Weiss, M.S. Global indicators of x-ray data quality. J. Appl Cryst. 2001, 34, 130–135. [Google Scholar] [CrossRef]
- Karplus, P.A.; Diederichs, K. Linking crystallographic model and data quality. Science 2012, 336, 1030–1033. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. Molprobity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Kleywegt, G.J.; Brunger, A.T. Checking your imagination: Applications of the free R value. Structure 1996, 4, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Cooper, D.R.; Porebski, P.J.; Shabalin, I.G.; Handing, K.B.; Minor, W. Checkmymetal: A macromolecular metal-binding validation tool. Acta Crystallogr. D Struct. Biol. 2017, 73, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Wedekind, J.E.; Trame, C.B.; Dorywalska, M.; Koehl, P.; Raschke, T.M.; McKee, M.; FitzGerald, D.; Collier, R.J.; McKay, D.B. Refined crystallographic structure of Pseudomonas aeruginosa exotoxin A and its implications for the molecular mechanism of toxicity. J. Mol. Biol. 2001, 314, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aqvist, J.; Luecke, H.; Quiocho, F.A.; Warshel, A. Dipoles localized at helix termini of proteins stabilize charges. Proc. Natl. Acad. Sci. USA 1991, 88, 2026–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedekind, J.E.; Liberman, J.A.; Jenkins, J.L.; Salim, M. Metal dependence of ligand binding and heavy-atom derivatization of evolutionarily distinct preQ1 riboswitches. In Chemical Biology of Nucleic Acids: Fundamentals and Clinical Applications; Erdmann, V., Markiewicz, W., Barciszewski, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 423–440. [Google Scholar]
- Wedekind, J.E. Metal ion binding and function in natural and artificial small RNA enzymes from a structural perspective. In Structural and Catalytic Roles of Metal Ions in RNA; Sigel, A., Sigel, H., Sigel, R., Eds.; Walter de Gruyter GmbH & Co KG: Berlin/Heidelberg, Germany, 2011; pp. 299–345. [Google Scholar]
- Bonn-Breach, R.; Gu, Y.; Jenkins, J.; Fasan, R.; Wedekind, J. Structure of sonic hedgehog protein in complex with zinc(II) and magnesium(II) reveals ion-coordination plasticity relevant to peptide drug design. Acta Crystallogr. D Struct. Biol. 2019, 75, 969–979. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Storoni, L.C.; Read, R.J. Simple algorithm for a maximum-likelihood SAD function. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Cromer, D.T. Anomalous dispersion corrections computed from self-consistent field relativistic Dirac–Slater wave functions. Acta Crystallogr. 1965, 18, 17–23. [Google Scholar] [CrossRef]
- Cromer, D.T.; Liberman, D. Relativistic calculation of anomalous scattering factors of x rays. J. Chem. Phys. 1970, 53, 1891–1898. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
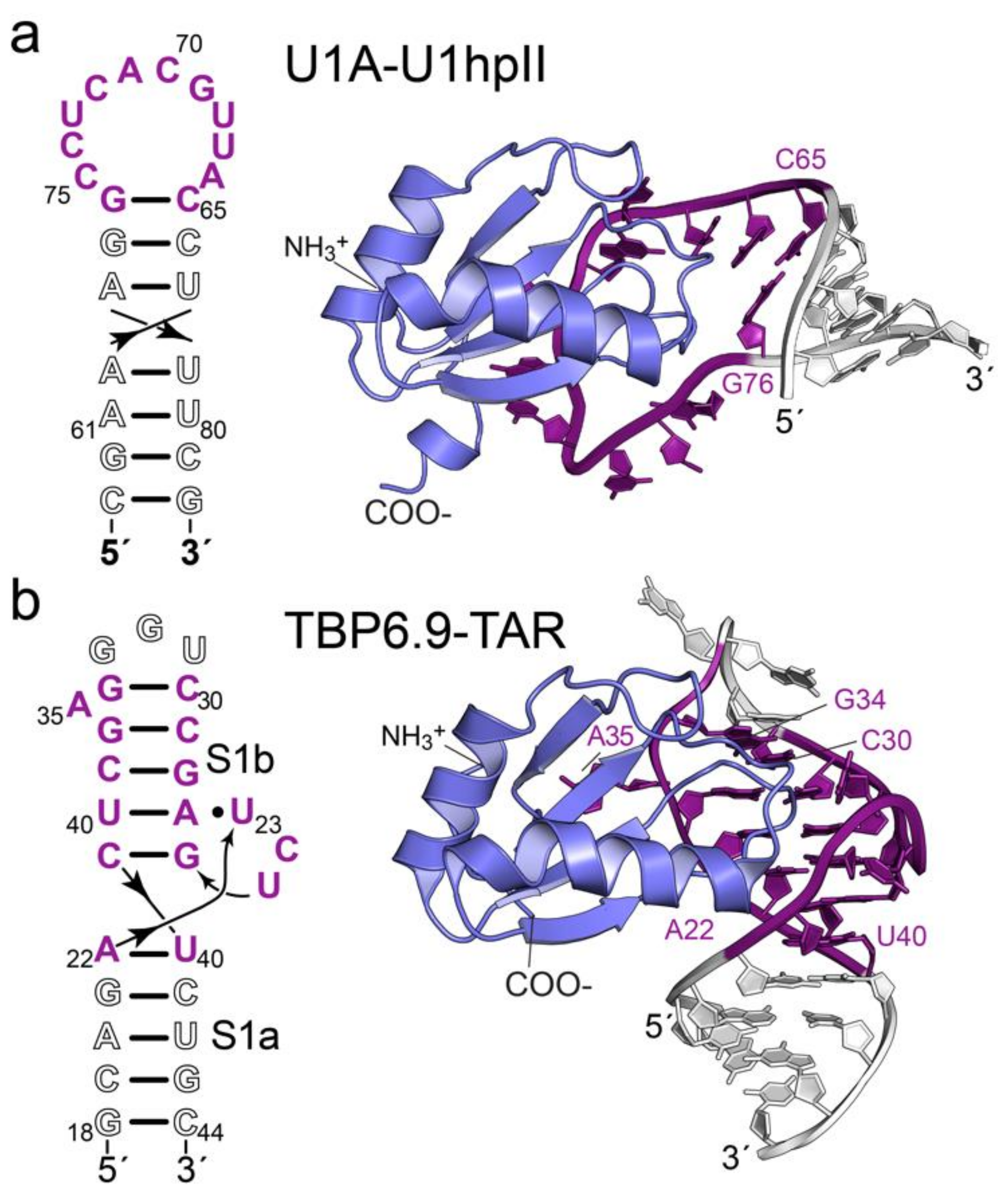{kind=link}
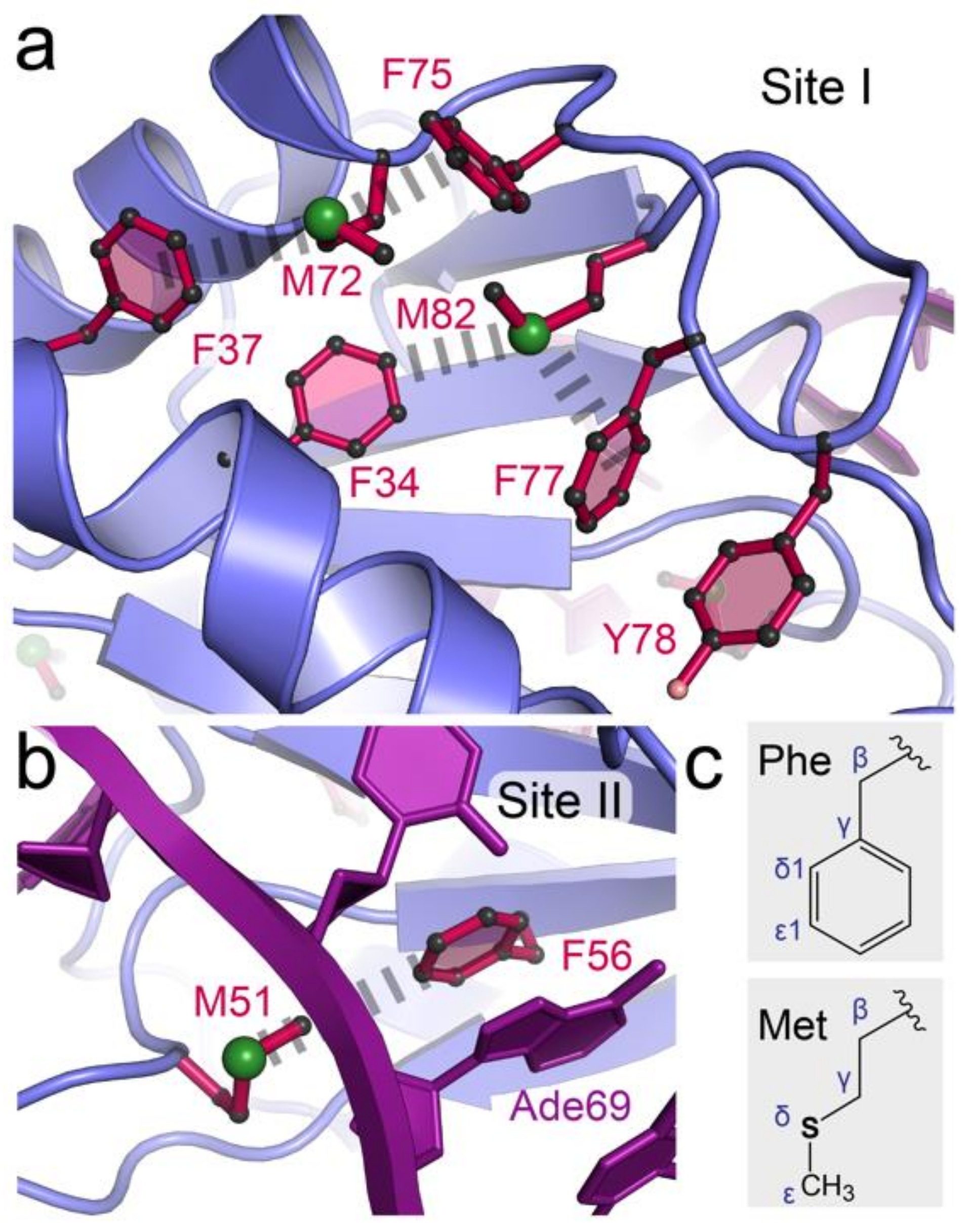{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | PDB ID | Phasing Method | No. Se Sites | Ref. |
|---|---|---|---|---|
| SAM-VI riboswitch | 6las | SAD a | 3–4 | [36] |
| NAD+-I riboswitch | 7d7v | SAD | 3 of 4 | [37] |
| HDV ribozyme | 1sjf | MAD b | 4 of 4 | [38] |
| HDV ribozyme | 1drz | MAD | 4 of 4 | [39] |
| group I intron | 1u6b | MAD c | 4 of 4 | [40] |
| glutamine riboswitch | 5ddp | MR d | n/a | [41] |
| glycine riboswitch | 3p49 | MAD e | n/a | [42] |
| cyclic-di-AMP riboswitch ydaO | 4w90 | MAD | 3 of 4 | [43] |
| c-di-GMP riboswitch | 3iwn | MR d | n/a | [44] |
| c-di-GMP riboswitch | 3irw | MAD e | n/a | [45] |
| aminoacyl tRNA synthetase ribozyme | 3cun | MAD/MR c | 3 of 4 | [46] |
| tetracycline artificial riboswitch | 3egz | MAD | 3 of 4 | [47] |
| class I ligase ribozyme | 3hhn | MAD e | n/a | [48] |
| pre-cleaved glmS ribozyme | 3g8S | MR d | n/a | [49] |
| K-turn-L7Ae complex | 4c4w | MR d | n/a | [50] |
| TAR RNA in complex with TAR binding protein 6.7 | 6cmn | MR f | n/a | [51] |
| TAR RNA in complex with TAR binding protein 6.9 | 6xh0 | MR g | n/a | [52] |
| Sample | KD nM | n | ∆H kcal mol−1 | −T∆S kcal mol−1 | ∆G kcal mol−1 | Krel a |
|---|---|---|---|---|---|---|
| dmU1A b | 152.0 ± 7.0 c | 1.0 ± 0.02 | −32.0 ± 0.6 | 22.8 ± 0.6 | −9.2 ± 0.03 | N/A |
| dmU1A 7Se-Met (F37M/F77M) b | 59.7 ± 11.4 | 0.95 ± 0.05 | −19.1 ± 3.0 | 9.4 ± 2.9 | −9.7 ± 0.12 | 0.39 |
| TBP6.9 (WT) d | 8.5 ± 0.4 | 1.0 ± 0.02 | −16.0 ± 0.1 | 5.2 ± 0.01 | −10.9 ± 0.1 | N/A |
| TBP6.9 7Se-Met (F34M/F37M/F77M) d | 13.5 ± 2.3 | 1.2 ± 0.02 | −14.5 ± 0.1 | 3.9 ± 0.1 | −10.6 ± 0.1 | 1.6 |
| TBP6.9 (WT) e | 87.8 ± 5.0 | 0.94 ± 0.04 | −7.8 ± 0.4 | −1.7 ± 0.5 | −9.5 ± 0.04 | 10.3 |
| TBP6.9 7Se-Met (F34M/F37M/F77M) e | 69.8 ± 2.9 | 1.1 ± 0.01 | −7.2 ± 0.8 | −2.4 ± 0.9 | −9.6 ± 0.03 | 8.2 |
| Data Collection | |
|---|---|
| Wavelength (Å) | 1.5418 |
| Resolution range (Å) | 27.88–2.20 (2.27–2.20) a |
| Space group | P 3 2 1 |
| a = b, c (Å) | 64.4, 46.8 |
| α = β, γ (°) | 90.0, 120 |
| Unique reflections | 10,961 |
| Multiplicity | 10.0 (4.8) a |
| Completeness (%) | 100.0 (100.0) a |
| Mean I/σ(I) | 61.2 (20.2) a |
| Rmerge (%) b | 2.9 (7.6) a |
| Rp.i.m. (%) c | 0.9 (3.8) a |
| CC1/2 (%) d | 1.00 (0.995) a |
| CCanom (%) e | 0.46 (0.10) a |
| RCRanom f | 1.65 (1.11) a |
| Refinement | |
| No. reflections (all/work/test) | 11519/10,961/558 |
| Rcryst/Rwork/Rfree (%) g | 16.8/16.6/21.3 |
| No. of atoms: | |
| protein | 776 |
| waters | 85 |
| Na/Acetate/βME | 13 |
| Se | 6 |
| R.M.S. deviations: | |
| bonds (Å) | 0.003 |
| angles (°) | 0.53 |
| ML coordinate error (Å) | 0.19 |
| Clashscore h | 0.0 |
| Molprobity scoreh | 0.50 |
| Ramachandran Plot (%): | |
| most favored | 100 |
| outliers (%) | 0 |
| B-factors overall (Å2): | 20.2 |
| protein | 19.6 |
| waters | 24.3 |
| Na/Acetate/βME | 30.8 |
| Se atoms | 27.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, Y.; Bonn-Breach, R.; Chavali, S.S.; Lippa, G.M.; Jenkins, J.L.; Wedekind, J.E. Affinity and Structural Analysis of the U1A RNA Recognition Motif with Engineered Methionines to Improve Experimental Phasing. Crystals 2021, 11, 273. https://doi.org/10.3390/cryst11030273
Srivastava Y, Bonn-Breach R, Chavali SS, Lippa GM, Jenkins JL, Wedekind JE. Affinity and Structural Analysis of the U1A RNA Recognition Motif with Engineered Methionines to Improve Experimental Phasing. Crystals. 2021; 11(3):273. https://doi.org/10.3390/cryst11030273
Chicago/Turabian StyleSrivastava, Yoshita, Rachel Bonn-Breach, Sai Shashank Chavali, Geoffrey M. Lippa, Jermaine L. Jenkins, and Joseph E. Wedekind. 2021. "Affinity and Structural Analysis of the U1A RNA Recognition Motif with Engineered Methionines to Improve Experimental Phasing" Crystals 11, no. 3: 273. https://doi.org/10.3390/cryst11030273
APA StyleSrivastava, Y., Bonn-Breach, R., Chavali, S. S., Lippa, G. M., Jenkins, J. L., & Wedekind, J. E. (2021). Affinity and Structural Analysis of the U1A RNA Recognition Motif with Engineered Methionines to Improve Experimental Phasing. Crystals, 11(3), 273. https://doi.org/10.3390/cryst11030273