Polymer@gold Nanoparticles Prepared via RAFT Polymerization for Opto-Biodetection




Abstract
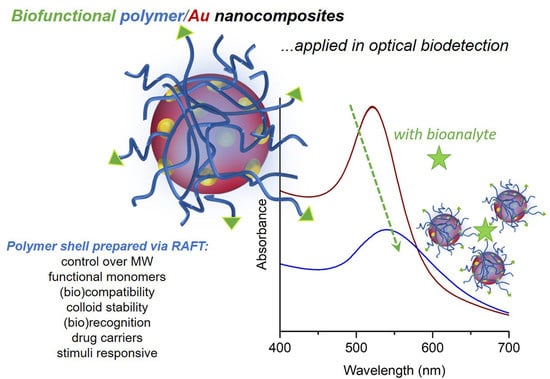
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
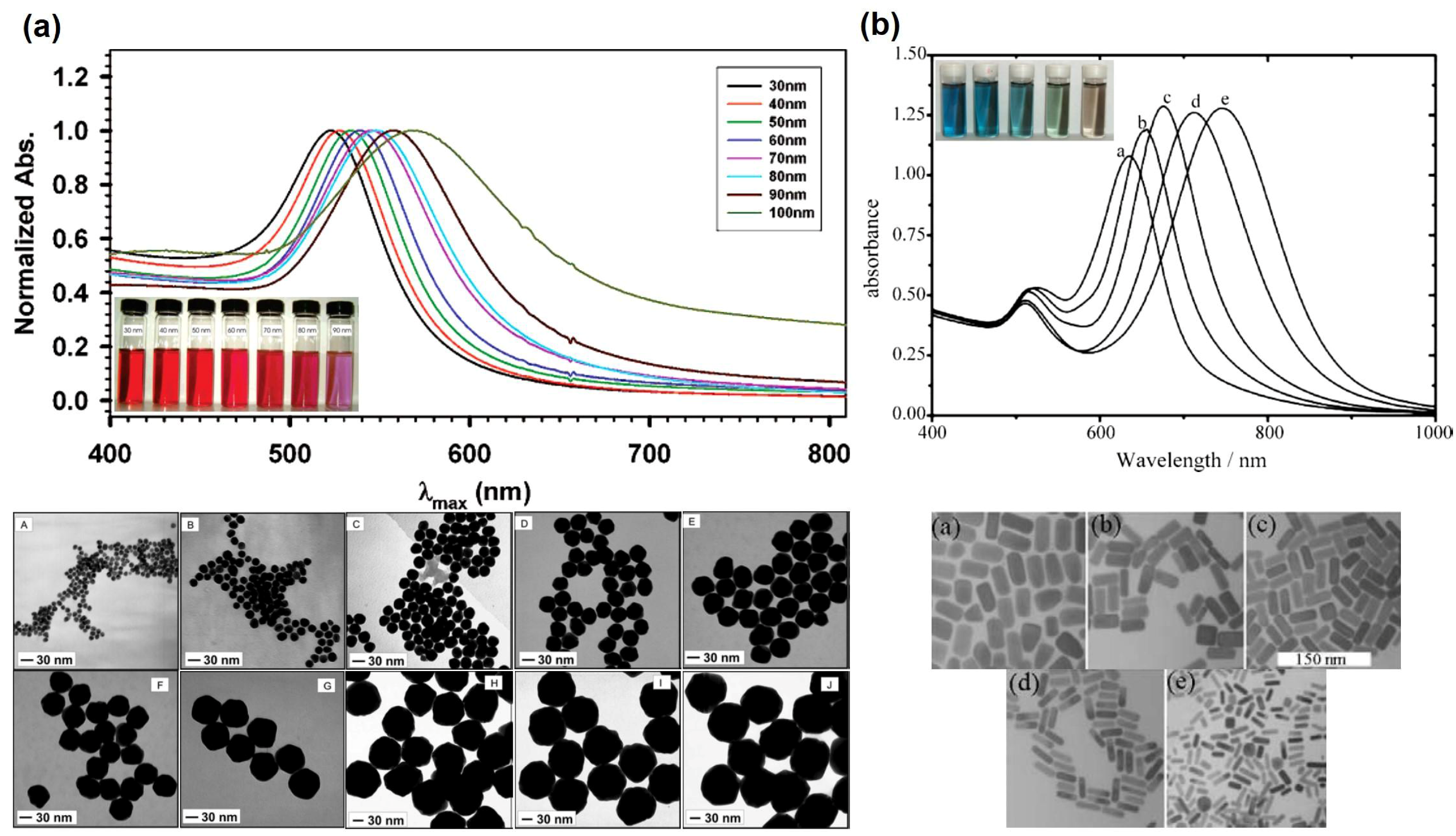
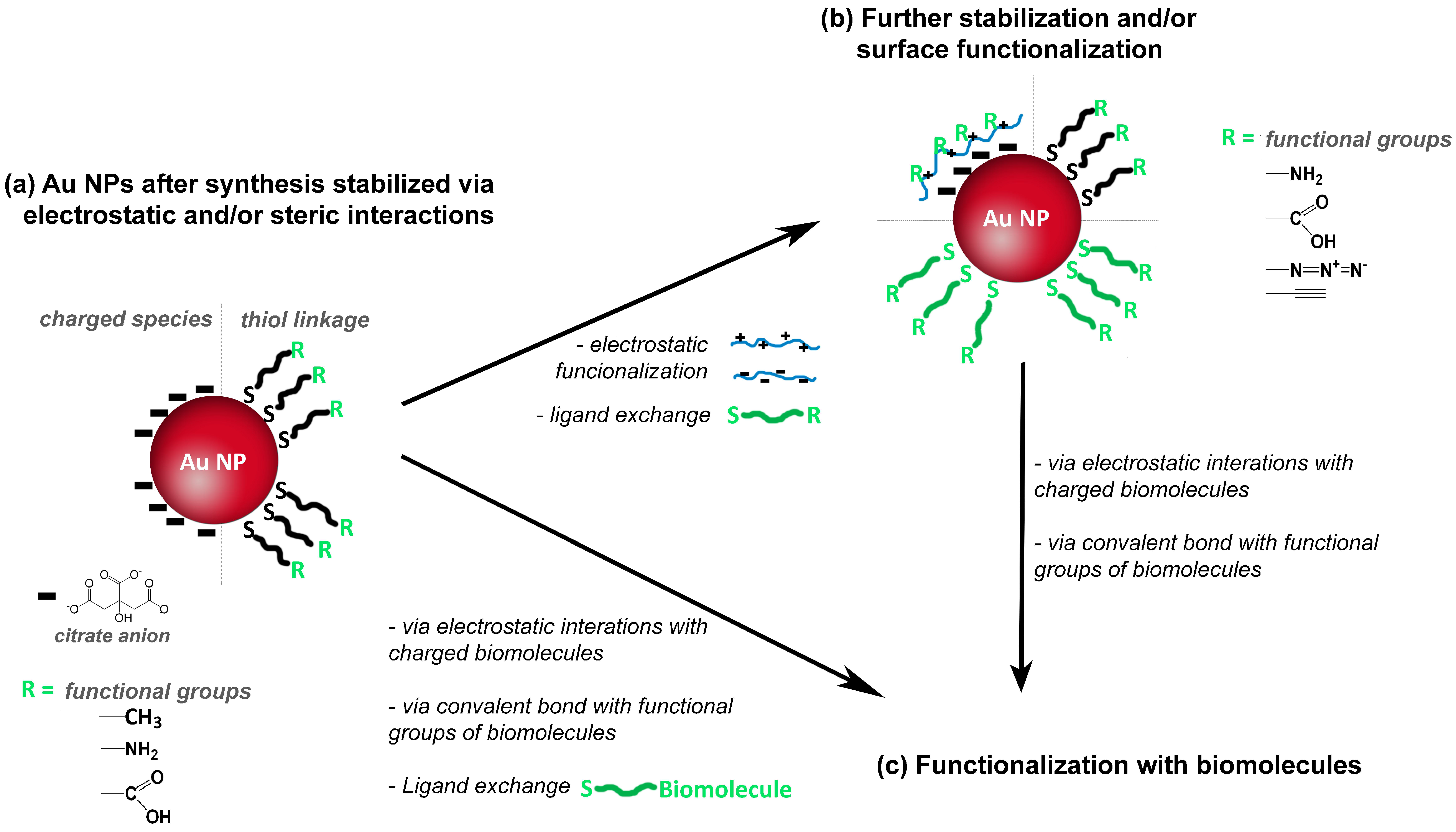
2. Colloidal Gold Nanoparticles: Optical Properties and Related Chemical Practices
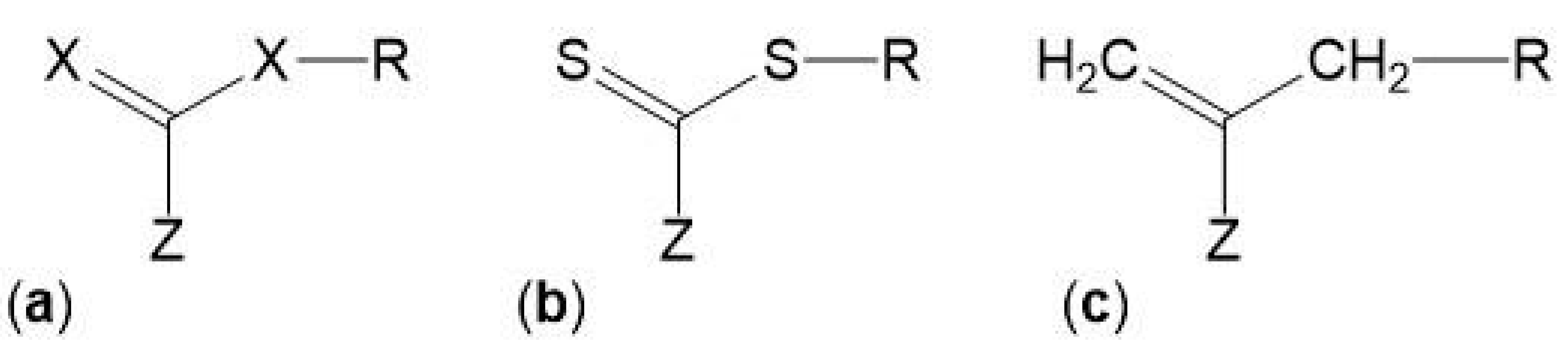
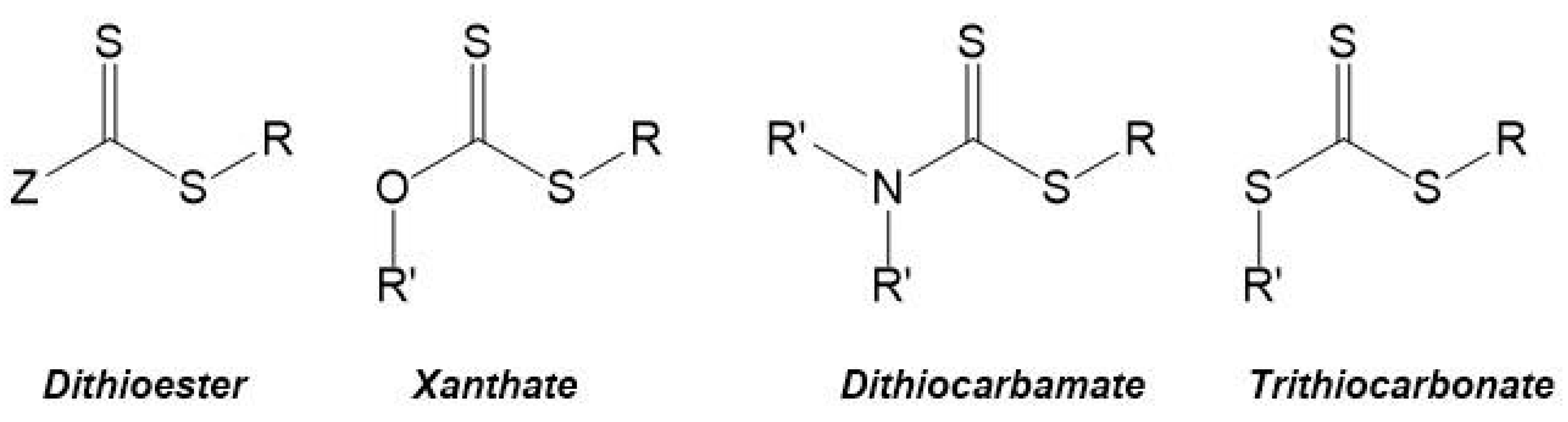
3. RAFT Polymerization
3.1. General Concepts
3.2. Application to Gold Nanocomposites
4. Au NPs in Opto-Biodetection and Beyond
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Beija, M.; Marty, J.-D.; Destarac, M. RAFT/MADIX Polymers for the Preparation of Polymer/inorganic Nanohybrids. Prog. Polym. Sci. 2011, 36, 845–886. [Google Scholar] [CrossRef]
- Liz-Marzán, L.M. Nanometals: Formation and Color. Mater. Today 2004, 7, 26–31. [Google Scholar] [CrossRef]
- Yeh, Y.-C.; Creran, B.; Rotello, V.M. Gold Nanoparticles: Preparation, Properties, and Applications in Bionanotechnology. Nanoscale 2012, 4, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Dreaden, E.C.; Alkilany, A.M.; Huang, X.; Murphy, C.J.; El-Sayed, M.A. The Golden Age: Gold Nanoparticles for Biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. [Google Scholar] [CrossRef] [PubMed]
- Doane, T.L.; Burda, C. The Unique Role of Nanoparticles in Nanomedicine: Imaging, Drug Delivery and Therapy. Chem. Soc. Rev. 2012, 41, 2885. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Neretina, S.; El-Sayed, M.A. Gold Nanorods: From Synthesis and Properties to Biological and Biomedical Applications. Adv. Mater. 2009, 21, 4880–4910. [Google Scholar] [CrossRef] [PubMed]
- Njoki, P.N.; Lim, I.-I.S.; Mott, D.; Park, H.-Y.; Khan, B.; Mishra, S.; Sujakumar, R.; Luo, J.; Zhong, C.-J. Size Correlation of Optical and Spectroscopic Properties for Gold Nanoparticles. J. Phys. Chem. C 2007, 111, 14664–14669. [Google Scholar] [CrossRef]
- Pérez-Juste, J.; Pastoriza-Santos, I.; Liz-Marzán, L.M.; Mulvaney, P. Gold Nanorods: Synthesis, Characterization and Applications. Coord. Chem. Rev. 2005, 249, 1870–1901. [Google Scholar] [CrossRef]
- Fateixa, S.; Correia, M.R.; Trindade, T. Resizing of Colloidal Gold Nanorods and Morphological Probing by SERS. J. Phys. Chem. C 2013, 117, 20343–20350. [Google Scholar] [CrossRef]
- Dulkeith, E.; Morteani, A.C.; Niedereichholz, T.; Klar, T.A.; Feldmann, J.; Levi, S.A.; van Veggel, F.C.; Reinhoudt, D.N.; Möller, M.; Gittins, D.I. Fluorescence Quenching of Dye Molecules near Gold Nanoparticles: Radiative and Nonradiative Effects. Phys. Rev. Lett. 2002, 89, 203002. [Google Scholar] [CrossRef] [PubMed]
- Gueroui, Z.; Libchaber, A. Single-Molecule Measurements of Gold-Quenched Quantum Dots. Phys. Rev. Lett. 2004, 93, 166108. [Google Scholar] [CrossRef] [PubMed]
- Jennings, T.L.; Singh, M.P.; Strouse, G.F. Fluorescent Lifetime Quenching near d = 1.5 nm Gold Nanoparticles: Probing NSET Validity. J. Am. Chem. Soc. 2006, 128, 5462–5467. [Google Scholar] [CrossRef] [PubMed]
- Sapsford, K.E.; Berti, L.; Medintz, I.L. Materials for Fluorescence Resonance Energy Transfer Analysis: Beyond Traditional Donor-Acceptor Combinations. Angew. Chem. Int. Ed. 2006, 45, 4562–4589. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.S.; Javier, A.; Jennings, T.; Fisher, M.; Hira, S.; Peterson, S.; Hopkins, B.; Reich, N.O.; Strouse, G.F. Nanometal Surface Energy Transfer in Optical Rulers, Breaking the FRET Barrier. J. Am. Chem. Soc. 2005, 127, 3115–3119. [Google Scholar] [CrossRef] [PubMed]
- Valeur, B. Molecular Fluorescence; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2001. [Google Scholar]
- Ray, P.C.; Darbha, G.K.; Ray, A.; Walker, J.; Hardy, W. Gold Nanoparticle Based FRET for DNA Detection. Plasmonics 2007, 2, 173–183. [Google Scholar] [CrossRef]
- Boisselier, E.; Astruc, D. Gold Nanoparticles in Nanomedicine: Preparations, Imaging, Diagnostics, Therapies and Toxicity. Chem. Soc. Rev. 2009, 38, 1759. [Google Scholar] [CrossRef] [PubMed]
- Doria, G.; Conde, J.; Veigas, B.; Giestas, L.; Almeida, C.; Assunção, M.; Rosa, J.; Baptista, P.V. Noble Metal Nanoparticles for Biosensing Applications. Sensors 2012, 12, 1657–1687. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.-C.; Astruc, D. Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications toward Biology, Catalysis, and Nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef] [PubMed]
- Dumur, F.; Guerlin, A.; Dumas, E.; Bertin, D.; Gigmes, D.; Mayer, C.R. Controlled Spontaneous Generation of Gold Nanoparticles Assisted by Dual Reducing and Capping Agents. Gold Bull. 2011, 44, 119–137. [Google Scholar] [CrossRef]
- Jimenez-Ruiz, A.; Perez-Tejeda, P.; Grueso, E.; Castillo, P.M.; Prado-Gotor, R. Nonfunctionalized Gold Nanoparticles: Synthetic Routes and Synthesis Condition Dependence. Chem. A Eur. J. 2015, 21, 9596–9609. [Google Scholar] [CrossRef] [PubMed]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A Study of the Nucleation and Growth Processes in the Synthesis of Colloidal Gold. Discuss. Faraday Soc. 1951, 11, 55–57. [Google Scholar] [CrossRef]
- Enustun, B.V.; Turkevich, J. Coagulation of Colloidal Gold. J. Am. Chem. Soc. 1963, 85, 3317–3328. [Google Scholar] [CrossRef]
- Park, J.W.; Shumaker-Parry, J.S. Structural Study of Citrate Layers on Gold Nanoparticles: Role of Intermolecular Interactions in Stabilizing Nanoparticles. J. Am. Chem. Soc. 2014, 136, 1907–1921. [Google Scholar] [CrossRef] [PubMed]
- Bastús, N.G.; Comenge, J.; Puntes, V. Kinetically Controlled Seeded Growth Synthesis of Citrate-Stabilized Gold Nanoparticles of up to 200 Nm: Size Focusing versus Ostwald Ripening. Langmuir 2011, 27, 11098–11105. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.; Eychmüller, A. Seeded Growth Synthesis of Uniform Gold Nanoparticles with Diameters of 15−300 Nm. J. Phys. Chem. C 2011, 115, 4502–4506. [Google Scholar] [CrossRef]
- Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D.J.; Whyman, R. Synthesis of Thiol-Derivatised Gold Nanoparticles in a Two-Phase Liquid?Liquid System. J. Chem. Soc. Chem. Commun. 1994, 801. [Google Scholar] [CrossRef]
- Perala, S.R.K.; Kumar, S. On the Mechanism of Metal Nanoparticle Synthesis in the Brust–Schiffrin Method. Langmuir 2013, 29, 9863–9873. [Google Scholar] [CrossRef] [PubMed]
- Gittins, D.I.; Caruso, F. Spontaneous Phase Transfer of Nanoparticulate Metals from Organic to Aqueous Media. Angew. Chem. Int. Ed. 2001, 40, 3001–3004. [Google Scholar] [CrossRef]
- Gandubert, V.J.; Lennox, R.B. Assessment of 4-(Dimethylamino)pyridine as a Capping Agent for Gold Nanoparticles. Langmuir 2005, 21, 6532–6539. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Graham, S.; Wang, Z.; Tan, B.; Sherrington, D.C.; Rannard, S.P.; Cooper, A.I.; Brust, M. Size-Controlled Synthesis of Near-Monodisperse Gold Nanoparticles in the 1−4 nm Range Using Polymeric Stabilizers. J. Am. Chem. Soc. 2005, 127, 16398–16399. [Google Scholar] [CrossRef] [PubMed]
- Lohse, S.E.; Dahl, J.A.; Hutchison, J.E. Direct Synthesis of Large Water-Soluble Functionalized Gold Nanoparticles Using Bunte Salts as Ligand Precursors. Langmuir 2010, 26, 7504–7511. [Google Scholar] [CrossRef] [PubMed]
- Jana, N.R.; Gearheart, L.; Murphy, C.J. Seeding Growth for Size Control of 5−40 nm Diameter Gold Nanoparticles. Langmuir 2001, 17, 6782–6786. [Google Scholar] [CrossRef]
- Sistach, S.; Rahme, K.; Pérignon, N.; Marty, J.-D.; Viguerie, N.L.; Gauffre, F.; Mingotaud, C. Bolaamphiphile Surfactants as Nanoparticle Stabilizers: Application to Reversible Aggregation of Gold Nanoparticles. Chem. Mater. 2008, 20, 1221–1223. [Google Scholar] [CrossRef]
- Keilitz, J.; Radowski, M.R.; Marty, J.; Haag, R.; Gauffre, F.; Mingotaud, C. Dendritic Polymers with a Core−Multishell Architecture: A Versatile Tool for the Stabilization of Nanoparticles. Chem. Mater. 2008, 20, 2423–2425. [Google Scholar] [CrossRef]
- Wang, S.; Qian, K.; Bi, X.; Huang, W. Influence of Speciation of Aqueous HAuCl4 on the Synthesis, Structure, and Property of Au Colloids. J. Phys. Chem. C 2009, 113, 6505–6510. [Google Scholar] [CrossRef]
- Ji, X.; Song, X.; Li, J.; Bai, Y.; Yang, W.; Peng, X. Size Control of Gold Nanocrystals in Citrate Reduction: The Third Role of Citrate. J. Am. Chem. Soc. 2007, 129, 13939–13948. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Jiang, D.; Cai, Y.; Ji, X.; Xie, R.; Yang, W. Tuning the Size of Gold Nanoparticles in the Citrate Reduction by Chloride Ions. Nanoscale 2012, 4, 5071. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.O.; Trindade, T.; Barros-Timmons, A. Impact of Critical Micelle Concentration of macroRAFT Agents on the Encapsulation of Colloidal Au Nanoparticles. Colloid Polym. Sci. 2018. submitted. [Google Scholar]
- Lohse, S.E.; Burrows, N.D.; Scarabelli, L.; Liz-Marzán, L.M.; Murphy, C.J. Anisotropic Noble Metal Nanocrystal Growth: The Role of Halides. Chem. Mater. 2014, 26, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Zhao, P.; Astruc, D. Anisotropic Gold Nanoparticles: Synthesis, Properties, Applications, and Toxicity. Angew. Chemie Int. Ed. 2014, 53, 1756–1789. [Google Scholar] [CrossRef] [PubMed]
- Jana, N.R.; Gearheart, L.; Murphy, C.J. Seed-Mediated Growth Approach for Shape-Controlled Synthesis of Spheroidal and Rod-like Gold Nanoparticles Using a Surfactant Template. Adv. Mater. 2001, 13, 1389–1393. [Google Scholar] [CrossRef]
- Jana, N.R.; Gearheart, L.; Murphy, C.J. Wet Chemical Synthesis of High Aspect Ratio Cylindrical Gold Nanorods. J. Phys. Chem. B 2001, 105, 4065–4067. [Google Scholar] [CrossRef]
- Gao, J.; Bender, C.M.; Murphy, C.J. Dependence of the Gold Nanorod Aspect Ratio on the Nature of the Directing Surfactant in Aqueous Solution. Langmuir 2003, 19, 9065–9070. [Google Scholar] [CrossRef]
- Nikoobakht, B.; El-Sayed, M.A. Preparation and Growth Mechanism of Gold Nanorods (NRs) Using Seed-Mediated Growth Method. Chem. Mater. 2003, 15, 1957–1962. [Google Scholar] [CrossRef]
- Gole, A.; Murphy, C.J. Seed-Mediated Synthesis of Gold Nanorods: Role of the Size and Nature of the Seed. Chem. Mater. 2004, 16, 3633–3640. [Google Scholar] [CrossRef]
- Murphy, C.J.; Thompson, L.B.; Chernak, D.J.; Yang, J.A.; Sivapalan, S.T.; Boulos, S.P.; Huang, J.; Alkilany, A.M.; Sisco, P.N. Gold Nanorod Crystal Growth: From Seed-Mediated Synthesis to Nanoscale Sculpting. Curr. Opin. Colloid Interface Sci. 2011, 16, 128–134. [Google Scholar] [CrossRef]
- Ye, X.; Gao, Y.; Chen, J.; Reifsnyder, D.C.; Zheng, C.; Murray, C.B. Seeded Growth of Monodisperse Gold Nanorods Using Bromide-Free Surfactant Mixtures. Nano Lett. 2013, 13, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Thanh, N.T.K.; Green, L.A.W. Functionalisation of Nanoparticles for Biomedical Applications. Nano Today 2010, 5, 213–230. [Google Scholar] [CrossRef]
- Cho, J.; Caruso, F. Investigation of the Interactions between Ligand-Stabilized Gold Nanoparticles and Polyelectrolyte Multilayer Films. Chem. Mater. 2005, 17, 4547–4553. [Google Scholar] [CrossRef]
- Gittins, D.I.; Caruso, F. Tailoring the Polyelectrolyte Coating of Metal Nanoparticles. J. Phys. Chem. B 2001, 105, 6846–6852. [Google Scholar] [CrossRef]
- Schneider, G.; Decher, G. From Functional Core/Shell Nanoparticles Prepared via Layer-by-Layer Deposition to Empty Nanospheres. Nano Lett. 2004, 4, 1833–1839. [Google Scholar] [CrossRef]
- Schneider, G.; Decher, G. Functional Core/shell Nanoparticles via Layer-by-Layer Assembly. Investigation of the Experimental Parameters for Controlling Particle Aggregation and for Enhancing Dispersion Stability. Langmuir 2008, 24, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Higashi, N.; Takagi, T.; Koga, T. Layer-by-Layer Fabrication of Well-Packed Gold Nanoparticle Assemblies Guided by a β-Sheet Peptide Network. Polym. J. 2010, 42, 95–99. [Google Scholar] [CrossRef]
- Beija, M.; Marty, J.-D.; Destarac, M. Thermoresponsive poly(N-Vinyl Caprolactam)-Coated Gold Nanoparticles: Sharp Reversible Response and Easy Tunability. Chem. Commun. 2011, 47, 2826. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Tenhu, H. Recent Advances in Polymer Protected Gold Nanoparticles: Synthesis, Properties and Applications. Chem. Commun. 2007, 4580–4598. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.O.; Barros-Timmons, A.; Trindade, T. Biofunctionalisation of Colloidal Gold Nanoparticles via Polyelectrolytes Assemblies. Colloid Polym. Sci. 2014, 292, 33–50. [Google Scholar] [CrossRef]
- Sato, K.; Hosokawa, K.; Maeda, M. Rapid Aggregation of Gold Nanoparticles Induced by Non-Cross-Linking DNA Hybridization. J. Am. Chem. Soc. 2003, 125, 8102–8103. [Google Scholar] [CrossRef] [PubMed]
- Zanoli, L.M.; D’Agata, R.; Spoto, G. Functionalized Gold Nanoparticles for Ultrasensitive DNA Detection. Anal. Bioanal. Chem. 2012, 402, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; He, Q.; Li, J. Smart Core/shell Nanocomposites: Intelligent Polymers Modified Gold Nanoparticles. Adv. Colloid Interface Sci. 2009, 149, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, P.K.; Ramani, K.P.; Singh, S.S.; Tekade, A.R.; Chatap, V.K.; Patil, G.B.; Bari, S.B. Stimuli-Sensitive Layer-by-Layer (LbL) Self-Assembly Systems: Targeting and Biosensory Applications. J. Control. Release 2013, 166, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.D.; Jones, R.G.; Moad, G. Terminology for Reversible-Deactivation Radical Polymerization Previously Called “controlled” radical Or “living” radical Polymerization (IUPAC Recommendations 2010). Pure Appl. Chem. 2009, 82, 483–491. [Google Scholar] [CrossRef]
- Cowie, J.M.G.; Arrighi, V. Polymers: Chemistry and Physics of Modern Materials, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living Radical Polymerization: Features, Developments, and Perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living Radical Polymerization by the RAFT Process. Aust. J. Chem. 2005, 58, 379. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living Radical Polymerization by the RAFT Process—A First Update. Aust. J. Chem. 2006, 59, 669. [Google Scholar] [CrossRef]
- York, A.; Kirkland, S.; McCormick, C. Advances in the Synthesis of Amphiphilic Block Copolymers via RAFT Polymerization: Stimuli-Responsive Drug and Gene Delivery. Adv. Drug Deliv. Rev. 2008, 60, 1018–1036. [Google Scholar] [CrossRef] [PubMed]
- Barner-Kowollik, C. Handbook of RAFT Polymerization; Wiley-VCH: Hoboken, NJ, USA, 2008. [Google Scholar]
- Moad, G.; Chiefari, J.; Chong, (Bill) Y.K.; Krstina, J.; Mayadunne, R.T.A.; Postma, A.; Rizzardo, E.; Thang, S.H. Living Free Radical Polymerization with Reversible Addition-fragmentation Chain Transfer (the Life of RAFT). Polym. Int. 2000, 49, 993–1001. [Google Scholar] [CrossRef]
- Lowe, A.B.; McCormick, C.L. Reversible Addition–fragmentation Chain Transfer (RAFT) Radical Polymerization and the Synthesis of Water-Soluble (Co)polymers under Homogeneous Conditions in Organic and Aqueous Media. Prog. Polym. Sci. 2007, 32, 283–351. [Google Scholar] [CrossRef]
- Favier, A.; Charreyre, M.-T. Experimental Requirements for an Efficient Control of Free-Radical Polymerizations via the Reversible Addition-Fragmentation Chain Transfer (RAFT) Process. Macromol. Rapid Commun. 2006, 27, 653–692. [Google Scholar] [CrossRef]
- Zapata-González, I.; Saldívar-Guerra, E.; Ortiz-Cisneros, J. Full Molecular Weight Distribution in RAFT Polymerization. New Mechanistic Insight by Direct Integration of the Equations. Macromol. Theory Simul. 2011, 20, 370–388. [Google Scholar] [CrossRef]
- Stenzel, M.H. Hairy Core-Shell Nanoparticles via RAFT: Where Are the Opportunities and Where Are the Problems and Challenges? Macromol. Rapid Commun. 2009, 30, 1603–1624. [Google Scholar] [CrossRef] [PubMed]
- Marien, Y.W.; Van Steenberge, P.H.M.; Kockler, K.B.; Barner-Kowollik, C.; Reyniers, M.-F.; D’hooge, D.R.; Marin, G.B. An Alternative Method to Estimate the Bulk Backbiting Rate Coefficient in Acrylate Radical Polymerization. Polym. Chem. 2016, 7, 6521–6528. [Google Scholar] [CrossRef]
- Derboven, P.; Van Steenberge, P.H.M.; Reyniers, M.-F.; Barner-Kowollik, C.; D’hooge, D.R.; Marin, G.B. Chain Transfer in Degenerative RAFT Polymerization Revisited: A Comparative Study of Literature Methods. Macromol. Theory Simul. 2016, 25, 104–115. [Google Scholar] [CrossRef]
- Marien, Y.W.; Van Steenberge, P.H.M.; Barner-Kowollik, C.; Reyniers, M.-F.; Marin, G.B.; D’hooge, D.R. Kinetic Monte Carlo Modeling Extracts Information on Chain Initiation and Termination from Complete PLP-SEC Traces. Macromolecules 2017, 50, 1371–1385. [Google Scholar] [CrossRef]
- Keddie, D.J. A Guide to the Synthesis of Block Copolymers Using Reversible-Addition Fragmentation Chain Transfer (RAFT) Polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Bourgeat-Lami, E.; França, A.J.P.G.; Chaparro, T.C.; Silva, R.D.; Dugas, P.-Y.; Alves, G.M.; Santos, A.M. Synthesis of Polymer/Silica Hybrid Latexes by Surfactant-Free RAFT-Mediated Emulsion Polymerization. Macromolecules 2016, 49, 4431–4440. [Google Scholar] [CrossRef]
- Gody, G.; Maschmeyer, T.; Zetterlund, P.B.; Perrier, S. Pushing the Limit of the RAFT Process: Multiblock Copolymers by One-Pot Rapid Multiple Chain Extensions at Full Monomer Conversion. Macromolecules 2014, 47, 3451–3460. [Google Scholar] [CrossRef]
- Sumerlin, B.S.; Lowe, A.B.; Stroud, P.A.; Zhang, P.; Urban, M.W.; McCormick, C.L. Modification of Gold Surfaces with Water-Soluble (Co)polymers Prepared via Aqueous Reversible Addition-fragmentation Chain Transfer (RAFT) Polymerization †. Langmuir 2003, 19, 5559–5562. [Google Scholar] [CrossRef]
- Duwez, A.-S.; Guillet, P.; Colard, C.; Gohy, J.-F.; Fustin, C.-A. Dithioesters and Trithiocarbonates as Anchoring Groups for the “Grafting-To” Approach. Macromolecules 2006, 39, 2729–2731. [Google Scholar] [CrossRef]
- Sze Ieong, N.; Biggs, C.I.; Walker, M.; Gibson, M.I. Comparison of RAFT-Derived Poly(vinylpyrrolidone) Verses Poly(oligoethyleneglycol Methacrylate) for the Stabilization of Glycosylated Gold Nanoparticles. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Kusolkamabot, K.; Sae-Ung, P.; Niamnont, N.; Wongravee, K.; Sukwattanasinitt, M.; Hoven, V.P. Poly(N-Isopropylacrylamide)-Stabilized Gold Nanoparticles in Combination with Tricationic Branched Phenylene-Ethynylene Fluorophore for Protein Identification. Langmuir 2013, 29, 12317–12327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Maji, S.; Antunes, A.B.D.F.; De Rycke, R.; Zhang, Q.; Hoogenboom, R.; De Geest, B.G. Salt Plays a Pivotal Role in the Temperature-Responsive Aggregation and Layer-by-Layer Assembly of Polymer-Decorated Gold Nanoparticles. Chem. Mater. 2013, 25, 4297–4303. [Google Scholar] [CrossRef]
- Durand-Gasselin, C.; Koerin, R.; Rieger, J.; Lequeux, N.; Sanson, N. Colloidal Stability of Zwitterionic Polymer-Grafted Gold Nanoparticles in Water. J. Colloid Interface Sci. 2014, 434, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Takara, M.; Toyoshima, M.; Seto, H.; Hoshino, Y.; Miura, Y. Polymer-Modified Gold Nanoparticles via RAFT Polymerization: A Detailed Study for a Biosensing Application. Polym. Chem. 2014, 5, 931–939. [Google Scholar] [CrossRef]
- Chen, N.; Xiang, X.; Heiden, P.A. Tuning Thermoresponsive Behavior of Diblock Copolymers and Their Gold Core Hybrids. Part 2. How Properties Change Depending on Block Attachment to Gold Nanoparticles. J. Colloid Interface Sci. 2013, 396, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Boyer, C.; Whittaker, M.R.; Chuah, K.; Liu, J.; Davis, T.P. Modulation of the Surface Charge on Polymer-Stabilized Gold Nanoparticles by the Application of an External Stimulus. Langmuir 2010, 26, 2721–2730. [Google Scholar] [CrossRef] [PubMed]
- Beija, M.; Palleau, E.; Sistach, S.; Zhao, X.; Ressier, L.; Mingotaud, C.; Destarac, M.; Marty, J.-D. Control of the Catalytic Properties and Directed Assembly on Surfaces of MADIX/RAFT Polymer-Coated Gold Nanoparticles by Tuning Polymeric Shell Charge. J. Mater. Chem. 2010, 20, 9433. [Google Scholar] [CrossRef]
- Gibson, M.I.; Paripovic, D.; Klok, H.-A. Size-Dependent LCST Transitions of Polymer-Coated Gold Nanoparticles: Cooperative Aggregation and Surface Assembly. Adv. Mater. 2010, 22, 4721–4725. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.I.; Danial, M.; Klok, H.-A. Sequentially Modified, Polymer-Stabilized Gold Nanoparticle Libraries: Convergent Synthesis and Aggregation Behavior. ACS Comb. Sci. 2011, 13, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, B.; Vana, P. RAFT-Polymers with Single and Multiple Trithiocarbonate Groups as Uniform Gold-Nanoparticle Coatings. Macromolecules 2013, 46, 4862–4871. [Google Scholar] [CrossRef]
- Rossner, C.; Ebeling, B.; Vana, P. Spherical Gold-Nanoparticle Assemblies with Tunable Interparticle Distances Mediated by Multifunctional RAFT Polymers. ACS Macro Lett. 2013, 2, 1073–1076. [Google Scholar] [CrossRef]
- Rossner, C.; Glatter, O.; Saldanha, O.; Köster, S.; Vana, P. The Structure of Gold-Nanoparticle Networks Cross-Linked by Di- and Multifunctional RAFT Oligomers. Langmuir 2015, 31, 10573–10582. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, J.W.; Lowe, A.B.; Boyes, S.G. Surface Modification of Gold Nanorods with Polymers Synthesized by Reversible Addition-fragmentation Chain Transfer Polymerization. Chem. Mater. 2007, 19, 6–13. [Google Scholar] [CrossRef]
- Sistach, S.; Beija, M.; Rahal, V.; Brûlet, A.; Marty, J.-D.; Destarac, M.; Mingotaud, C. Thermoresponsive Amphiphilic Diblock Copolymers Synthesized by MADIX/RAFT: Properties in Aqueous Solutions and Use for the Preparation and Stabilization of Gold Nanoparticles. Chem. Mater. 2010, 22, 3712–3724. [Google Scholar] [CrossRef]
- Pereira, S.O.; Barros-Timmons, A.; Trindade, T. A Comparative Study of Chemical Routes for Coating Gold Nanoparticles via Controlled RAFT Emulsion Polymerization. Part. Part. Syst. Charact. 2017. [Google Scholar] [CrossRef]
- Boyer, C.; Whittaker, M.R.; Nouvel, C.; Davis, T.P. Synthesis of Hollow Polymer Nanocapsules Exploiting Gold Nanoparticles as Sacrificial Templates. Macromolecules 2010, 43, 1792–1799. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X. Recent Advances in Block Copolymer-Assisted Synthesis of Supramolecular Inorganic/organic Hybrid Colloids. Polym. Chem. 2011, 2, 2741. [Google Scholar] [CrossRef]
- Grzelczak, M.; Sánchez-Iglesias, A.; Liz-Marzán, L.M. A General Approach toward Polymer-Coated Plasmonic Nanostructures. CrystEngComm 2014, 16, 9425–9429. [Google Scholar] [CrossRef]
- Lowe, A.B.; Sumerlin, B.S.; Donovan, M.S.; McCormick, C.L. Facile Preparation of Transition Metal Nanoparticles Stabilized by Well-Defined (Co)polymers Synthesized via Aqueous Reversible Addition-Fragmentation Chain Transfer Polymerization. J. Am. Chem. Soc. 2002, 124, 11562–11563. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.J.; Go, D.H.; Choi, S.; Kim, K.M.; Lee, J.Y.; Choo, D.J.; Yoo, H.-O.; Kim, J.M.; Kim, J. Synthesis of Poly(ethylene Oxide)-Based Thermoresponsive Block Copolymers by RAFT Radical Polymerization and Their Uses for Preparation of Gold Nanoparticles. Colloids Surfaces A Physicochem. Eng. Asp. 2008, 317, 496–503. [Google Scholar] [CrossRef]
- Wang, Z.L.; Xu, J.T.; Du, B.Y.; Fan, Z.Q. Facile Fabrication of Amphiphilic Gold Nanoparticles with V-Shaped Brushes from Block Copolymers with a Trithiocarbonate Group as the Junction. J. Colloid Interface Sci. 2011, 360, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Glaria, A.; Beija, M.; Bordes, R.; Destarac, M.; Marty, J.-D. Understanding the Role of ω-End Groups and Molecular Weight in the Interaction of PNIPAM with Gold Surfaces. Chem. Mater. 2013, 25, 1868–1876. [Google Scholar] [CrossRef]
- Wu, L.; Glebe, U.; Böker, A. Surface-Initiated Controlled Radical Polymerizations from Silica Nanoparticles, Gold Nanocrystals, and Bionanoparticles. Polym. Chem. 2015, 6, 5143–5184. [Google Scholar] [CrossRef]
- Raula, J.; Shan, J.; Nuopponen, M.; Niskanen, A.; Jiang, H.; Kauppinen, E.I.; Tenhu, H. Synthesis of Gold Nanoparticles Grafted with a Thermoresponsive Polymer by Surface-Induced Reversible-Addition-Fragmentation Chain-Transfer Polymerization. Langmuir 2003, 19, 3499–3504. [Google Scholar] [CrossRef]
- Esteves, A.C.C.; Hodge, P.; Trindade, T.; Barros-Timmons, A.M.M.V. Preparation of Nanocomposites by Reversible Addition-Fragmentation Chain Transfer Polymerization from the Surface of Quantum Dots in Miniemulsion. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 5367–5377. [Google Scholar] [CrossRef]
- Nguyen, D.; Zondanos, H.S.; Farrugia, J.M.; Serelis, A.K.; Such, C.H.; Hawkett, B.S. Pigment Encapsulation by Emulsion Polymerization Using Macro-RAFT Copolymers. Langmuir 2008, 24, 2140–2150. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.I.; Heuts, J.P.A.; Hawkett, B.S.; van Herk, A.M. Polymer Encapsulated Gibbsite Nanoparticles: Efficient Preparation of Anisotropic Composite Latex Particles by RAFT-Based Starved Feed Emulsion Polymerization. Langmuir 2009, 25, 10523–10533. [Google Scholar] [CrossRef] [PubMed]
- Mballa Mballa, M.A.; Ali, S.I.; Heuts, J.P.; van Herk, A.M. Control of the Anisotropic Morphology of Latex Nanocomposites Containing Single Montmorillonite Clay Particles Prepared by Conventional and Reversible Addition-Fragmentation Chain Transfer Based Emulsion Polymerization. Polym. Int. 2012, 61, 861–865. [Google Scholar] [CrossRef]
- Perreira, A.C.; Pearson, S.; Kostadinova, D.; Leroux, F.; D’Agosto, F.; Lansalot, M.; Bourgeat-Lami, E.; Prévot, V. Nanocomposite Latexes Containing Layered Double Hydroxides via RAFT-Assisted Encapsulating Emulsion Polymerization. Polym. Chem. 2017, 8, 1233–1243. [Google Scholar] [CrossRef]
- Zhong, W.; Zeuna, J.N.; Claverie, J.P. A Versatile Encapsulation Method of Noncovalently Modified Carbon Nanotubes by RAFT Polymerization. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 4403–4407. [Google Scholar] [CrossRef]
- Nguyen, D.; Such, C.H.; Hawkett, B.S. Polymer Coating of Carboxylic Acid Functionalized Multiwalled Carbon Nanotubes via Reversible Addition-Fragmentation Chain Transfer Mediated Emulsion Polymerization. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 250–257. [Google Scholar] [CrossRef]
- Huynh, V.T.; Nguyen, D.; Such, C.H.; Hawkett, B.S. Polymer Coating of Graphene Oxide via Reversible Addition-Fragmentation Chain Transfer Mediated Emulsion Polymerization. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 1413–1421. [Google Scholar] [CrossRef]
- Garnier, J.; Warnant, J.; Lacroix-Desmazes, P.; Dufils, P.-E.; Vinas, J.; Vanderveken, Y.; van Herk, A.M. An Emulsifier-Free RAFT-Mediated Process for the Efficient Synthesis of Cerium Oxide/Polymer Hybrid Latexes. Macromol. Rapid Commun. 2012, 33, 1388–1392. [Google Scholar] [CrossRef] [PubMed]
- Zgheib, N.; Putaux, J.-L.; Thill, A.; Bourgeat-Lami, E.; D’Agosto, F.; Lansalot, M. Cerium Oxide Encapsulation by Emulsion Polymerization Using Hydrophilic macroRAFT Agents. Polym. Chem. 2013, 4, 607–614. [Google Scholar] [CrossRef]
- Garnier, J.; Warnant, J.; Lacroix-Desmazes, P.; Dufils, P.-E.; Vinas, J.; van Herk, A. Sulfonated Macro-RAFT Agents for the Surfactant-Free Synthesis of Cerium Oxide-Based Hybrid Latexes. J. Colloid Interface Sci. 2013, 407, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Zhong, W.; Claverie, J.P. Copolymer Nanosphere Encapsulated CdS Quantum Dots Prepared by RAFT Copolymerization: Synthesis, Characterization and Mechanism of Formation. Colloid Polym. Sci. 2011, 289, 1519–1533. [Google Scholar] [CrossRef]
- Das, P.; Claverie, J.P. Synthesis of Single-Core and Multiple-Core Core-Shell Nanoparticles by RAFT Emulsion Polymerization: Lead Sulfide-Copolymer Nanocomposites. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 2802–2808. [Google Scholar] [CrossRef]
- Warnant, J.; Garnier, J.; van Herk, A.; Dufils, P.-E.; Vinas, J.; Lacroix-Desmazes, P. A CeO2/PVDC Hybrid Latex Mediated by a Phosphonated Macro-RAFT Agent. Polym. Chem. 2013, 4, 5656. [Google Scholar] [CrossRef]
- Silva, R.D.; Stefanichen Monteiro, I.; Chaparro, T.D.C.; Silva Hardt, R.; Giudici, R.; Barros-Timmons, A.; Bourgeat-Lami, E.; Martins dos Santos, A. Investigation of the Adsorption of Amphipathic macroRAFT Agents onto Montmorillonite Clay. Langmuir 2017, 33, 9598–9608. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Melro, L.; de Camargo Chaparro, T.; de Souza Filho, I.R.; Ananias, D.; Bourgeat-Lami, E.; dos Santos, A.M.; Barros-Timmons, A. Adsorption Study of a Macro-RAFT Agent onto SiO2-Coated Gd2O3:Eu3+ Nanorods: Requirements and Limitations. Appl. Surf. Sci. 2017, 394, 519–527. [Google Scholar] [CrossRef]
- Saha, K.; Agasti, S.S.; Kim, C.; Li, X.; Rotello, V.M. Gold Nanoparticles in Chemical and Biological Sensing. Chem. Rev. 2012, 112, 2739–2779. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Yong, K.-T.; Roy, I.; Dinh, X.-Q.; Yu, X.; Luan, F. A Review on Functionalized Gold Nanoparticles for Biosensing Applications. Plasmonics 2011, 6, 491–506. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Y.; Chen, L.; Choo, J. Nanomaterial-Assisted Aptamers for Optical Sensing. Biosens. Bioelectron. 2010, 25, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R. The Use of Gold Nanoparticles in Diagnostics and Detection. Chem. Soc. Rev. 2008, 37, 2028–2045. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Hu, C.-C.; Chang, H.-T.; Lu, C. Gold Nanoparticles as Sensitive Optical Probes. Analyst 2016, 141, 1611–1626. [Google Scholar] [CrossRef] [PubMed]
- Storhoff, J.J.; Lazarides, A.A.; Mucic, R.C.; Mirkin, C.A.; Letsinger, R.L.; Schatz, G.C. What Controls the Optical Properties of DNA-Linked Gold Nanoparticle Assemblies? J. Am. Chem. Soc. 2000, 122, 4640–4650. [Google Scholar] [CrossRef]
- Kohut, A.; Voronov, A.; Peukert, W. Organization of Functionalized Gold Nanoparticles by Controlled Protein Interactions. Part. Part. Syst. Charact. 2005, 22, 329–335. [Google Scholar] [CrossRef]
- Aslan, K.; Luhrs, C.C.; Pérez-Luna, V.H. Controlled and Reversible Aggregation of Biotinylated Gold Nanoparticles with Streptavidin. J. Phys. Chem. B 2004, 108, 15631–15639. [Google Scholar] [CrossRef]
- Pereira, S.O.; Trindade, T.; Barros-Timmons, A. Biotinylation of Optically Responsive Gold/polyelectrolyte Nanostructures. Gold Bull. 2015, 48. [Google Scholar] [CrossRef]
- Kato, N.; Caruso, F. Homogeneous, Competitive Fluorescence Quenching Immunoassay Based on Gold Nanoparticle/polyelectrolyte Coated Latex Particles. J. Phys. Chem. B 2005, 109, 19604–19612. [Google Scholar] [CrossRef] [PubMed]
- Aslan, K.; Pérez-Luna, V.H. Nonradiative Interactions between Biotin-Functionalized Gold Nanoparticles and Fluorophore-Labeled Antibiotin. Plasmonics 2006, 1, 111–119. [Google Scholar] [CrossRef]
- Dubertret, B.; Calame, M.; Libchaber, A.J. Single-Mismatch Detection Using Gold-Quenched Fluorescent Oligonucleotides. Nat. Biotechnol. 2001, 19, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Obliosca, J.M.; Wang, P.-C.; Tseng, F.-G. Probing Quenched Dye Fluorescence of Cy3-DNA-Au-Nanoparticle Hybrid Conjugates Using Solution and Array Platforms. J. Colloid Interface Sci. 2012, 371, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Aslan, K.; Pérez-Luna, V.H. Quenched Emission of Fluorescence by Ligand Functionalized Gold Nanoparticles. J. Fluoresc. 2004, 14, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Paino, M.; Bordegé, V.; Cuervo-Rodríguez, R.; Muñoz-Bonilla, A.; Fernández-García, M. Well-Defined Glycopolymers via RAFT Polymerization: Stabilization of Gold Nanoparticles. Macromol. Chem. Phys. 2014, 215, 1915–1924. [Google Scholar] [CrossRef]
- Zhang, Z.; Schepens, B.; Nuhn, L.; Saelens, X.; Schotsaert, M.; Callewaert, N.; De Rycke, R.; Zhang, Q.; Moins, S.; Benali, S.; et al. Influenza-Binding Sialylated Polymer Coated Gold Nanoparticles Prepared via RAFT Polymerization and Reductive Amination. Chem. Commun. 2016, 52, 3352–3355. [Google Scholar] [CrossRef] [PubMed]
- Parry, A.L.; Clemson, N.A.; Ellis, J.; Bernhard, S.S.R.; Davis, B.G.; Cameron, N.R. “Multicopy Multivalent” Glycopolymer-Stabilized Gold Nanoparticles as Potential Synthetic Cancer Vaccines. J. Am. Chem. Soc. 2013, 135, 9362–9365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kneipp, J.; Kneipp, H.; Kneipp, K. SERS—A Single-Molecule and Nanoscale Tool for Bioanalytics. Chem. Soc. Rev. 2008, 37, 1052. [Google Scholar] [CrossRef] [PubMed]
- Strozyk, M.S.; Jimenez de Aberasturi, D.; Liz-Marzán, L.M. Composite Polymer Colloids for SERS-Based Applications. Chem. Rec. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fateixa, S.; Nogueira, H.I.S.; Trindade, T. Hybrid Nanostructures for SERS: Materials Development and Chemical Detection. Phys. Chem. Chem. Phys. 2015, 17, 21046–21071. [Google Scholar] [CrossRef] [PubMed]
- Schiller, T.L.; Keddie, D.J.; Blakey, I.; Fredericks, P.M. Surface-Enhanced Raman Encoded Polymer Stabilized Gold Nanoparticles: Demonstration of Potential for Use in Bioassays. Eur. Polym. J. 2017, 87, 508–518. [Google Scholar] [CrossRef]
- Dey, P.; Blakey, I.; Thurecht, K.J.; Fredericks, P.M. Self-Assembled Hyperbranched Polymer–Gold Nanoparticle Hybrids: Understanding the Effect of Polymer Coverage on Assembly Size and SERS Performance. Langmuir 2013, 29, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Cesur, B.; Zhang, Z.; De Geest, B.G.; Hoogenboom, R. Poly(N-Isopropylacrylamide) Coated Gold Nanoparticles as Colourimetric Temperature and Salt Sensors. Polym. Chem. 2016, 7, 1705–1710. [Google Scholar] [CrossRef]
- Han, F.; Soeriyadi, A.H.; Vivekchand, S.R.C.; Gooding, J.J. Simple Method for Tuning the Optical Properties of Thermoresponsive Plasmonic Nanogels. ACS Macro Lett. 2016, 5, 626–630. [Google Scholar] [CrossRef]
- Jones, S.T.; Walsh-Korb, Z.; Barrow, S.J.; Henderson, S.L.; del Barrio, J.; Scherman, O.A. The Importance of Excess Poly(N-Isopropylacrylamide) for the Aggregation of Poly(N-Isopropylacrylamide)-Coated Gold Nanoparticles. ACS Nano 2016, 10, 3158–3165. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Phillips, D.J.; Walker, M.; Gibson, M.I. Co-Operative Transitions of Responsive-Polymer Coated Gold Nanoparticles; Precision Tuning and Direct Evidence for Co-Operative Aggregation. J. Mater. Chem. B 2016, 4, 5673–5682. [Google Scholar] [CrossRef] [PubMed]
- Boyer, C.; Whittaker, M.R.; Luzon, M.; Davis, T.P. Design and Synthesis of Dual Thermoresponsive and Antifouling Hybrid Polymer/Gold Nanoparticles. Macromolecules 2009, 42, 6917–6926. [Google Scholar] [CrossRef]
- Chen, N.; Xiang, X.; Tiwari, A.; Heiden, P.A. Tuning Thermoresponsive Behavior of Diblock Copolymers and Their Gold Core Hybrids. Part 1. Importance of Placement of Amphiphilic End Groups on the Diblock Copolymers. J. Colloid Interface Sci. 2013, 391, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Zhang, Z.; Voorhaar, L.; Pieters, S.; Stubbe, B.; Van Vlierberghe, S.; Dubruel, P.; De Geest, B.G.; Hoogenboom, R. Thermoresponsive Polymer Coated Gold Nanoparticles: From MADIX/RAFT Copolymerization of N-Vinylpyrrolidone and N-Vinylcaprolactam to Salt and Temperature Induced Nanoparticle Aggregation. RSC Adv. 2015, 5, 42388–42398. [Google Scholar] [CrossRef]
- Cortez-Lemus, N.A.; Licea-Claverie, A. RAFT Synthesis of poly(2-Dimethylaminoethyl Methacrylate) Three-Arm Star Polymers for the Preparation of Gold Nanoparticles. Polym. Bull. 2014, 71, 1757–1772. [Google Scholar] [CrossRef]
- Honold, T.; Skrybeck, D.; Wagner, K.G.; Karg, M. Fully Reversible Quantitative Phase Transfer of Gold Nanoparticles Using Bifunctional PNIPAM Ligands. Langmuir 2017, 33, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Abadeer, N.S.; Murphy, C.J. Recent Progress in Cancer Thermal Therapy Using Gold Nanoparticles. J. Phys. Chem. C 2016, 120, 4691–4716. [Google Scholar] [CrossRef]
- Gharatape, A.; Salehi, R. Recent Progress in Theranostic Applications of Hybrid Gold Nanoparticles. Eur. J. Med. Chem. 2017, 138, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Guler, E.; Geyik, C.; Demir, B.; Ozkan, M.; Odaci Demirkol, D.; Ozcelik, S.; Timur, S.; Becer, C.R. pH Responsive Glycopolymer Nanoparticles for Targeted Delivery of Anti-Cancer Drugs. Mol. Syst. Des. Eng. 2018. [Google Scholar] [CrossRef]
- Huang, X.; Jain, P.K.; El-Sayed, I.H.; El-Sayed, M.A. Plasmonic Photothermal Therapy (PPTT) Using Gold Nanoparticles. Lasers Med. Sci. 2008, 23, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Detrembleur, C.; De Pauw-Gillet, M.-C.; Mornet, S.; Duguet, E.; Jérôme, C. Gold Nanorods Coated with a Thermo-Responsive Poly(ethylene Glycol)-B-poly(N-Vinylcaprolactam) Corona as Drug Delivery Systems for Remotely near Infrared-Triggered Release. Polym. Chem. 2014, 5, 799–813. [Google Scholar] [CrossRef]
- Lin, I.-C.; Liang, M.; Liu, T.-Y.; Ziora, Z.M.; Monteiro, M.J.; Toth, I. Interaction of Densely Polymer-Coated Gold Nanoparticles with Epithelial Caco-2 Monolayers. Biomacromolecules 2011, 12, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Lin, I.-C.; Whittaker, M.R.; Minchin, R.F.; Monteiro, M.J.; Toth, I. Cellular Uptake of Densely Packed Polymer Coatings on Gold Nanoparticles. ACS Nano 2010, 4, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lawrence, J.; Parelkar, S.; Emrick, T. Novel Zwitterionic Copolymers with Dihydrolipoic Acid: Synthesis and Preparation of Nonfouling Nanorods. Macromolecules 2013, 46, 119–127. [Google Scholar] [CrossRef]













© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, S.O.; Barros-Timmons, A.; Trindade, T. Polymer@gold Nanoparticles Prepared via RAFT Polymerization for Opto-Biodetection. Polymers 2018, 10, 189. https://doi.org/10.3390/polym10020189
Pereira SO, Barros-Timmons A, Trindade T. Polymer@gold Nanoparticles Prepared via RAFT Polymerization for Opto-Biodetection. Polymers. 2018; 10(2):189. https://doi.org/10.3390/polym10020189
Chicago/Turabian StylePereira, Sónia O., Ana Barros-Timmons, and Tito Trindade. 2018. "Polymer@gold Nanoparticles Prepared via RAFT Polymerization for Opto-Biodetection" Polymers 10, no. 2: 189. https://doi.org/10.3390/polym10020189
APA StylePereira, S. O., Barros-Timmons, A., & Trindade, T. (2018). Polymer@gold Nanoparticles Prepared via RAFT Polymerization for Opto-Biodetection. Polymers, 10(2), 189. https://doi.org/10.3390/polym10020189