
Hg(II) Coordination Polymers Based on N,N’-bis(pyridine-4-yl)formamidine



Abstract
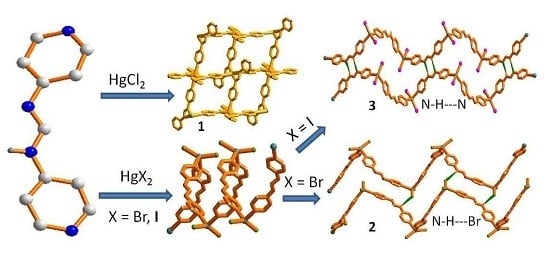
:
1. Introduction
2. Experimental Section
2.1. General Procedures
2.2. Materials
2.3. Preparations
2.3.1. Synthesis of 4-Hpyf
2.3.2. Synthesis of {[Hg(4-pyf)2]·2(THF)}n, 1
2.3.3. Synthesis of {[HgBr2(4-Hpyf)]·(CH3CN)}n, 2, and {[HgI2(4-Hpyf)]·(CH3CN)}n, 3
2.4. X-ray Crystallography
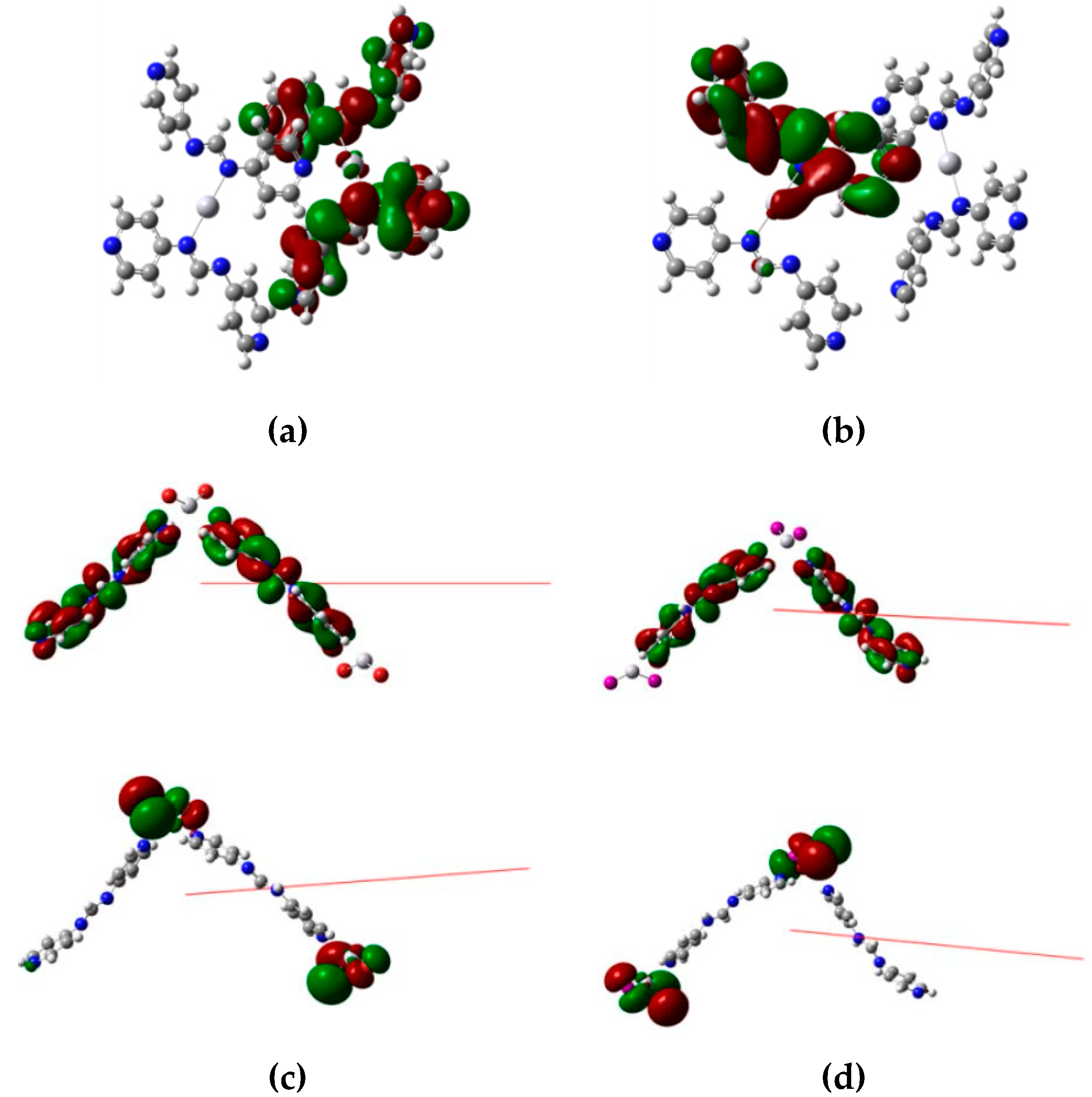
2.5. Computational Methods
2.6. Gas Adsorption Measurements
3. Results and Discussion
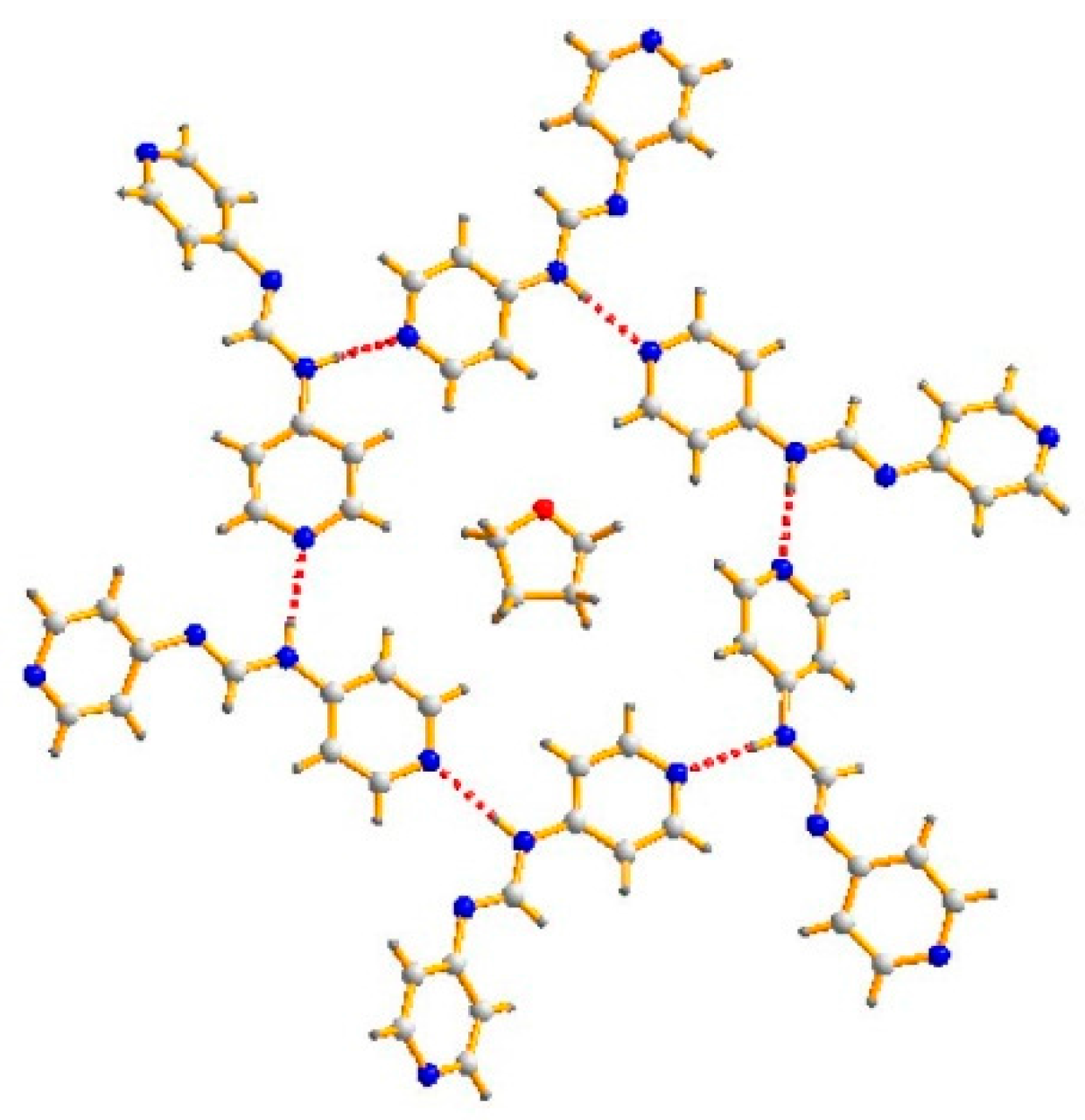
3.1. Structure of 4-Hpyf·0.16THF
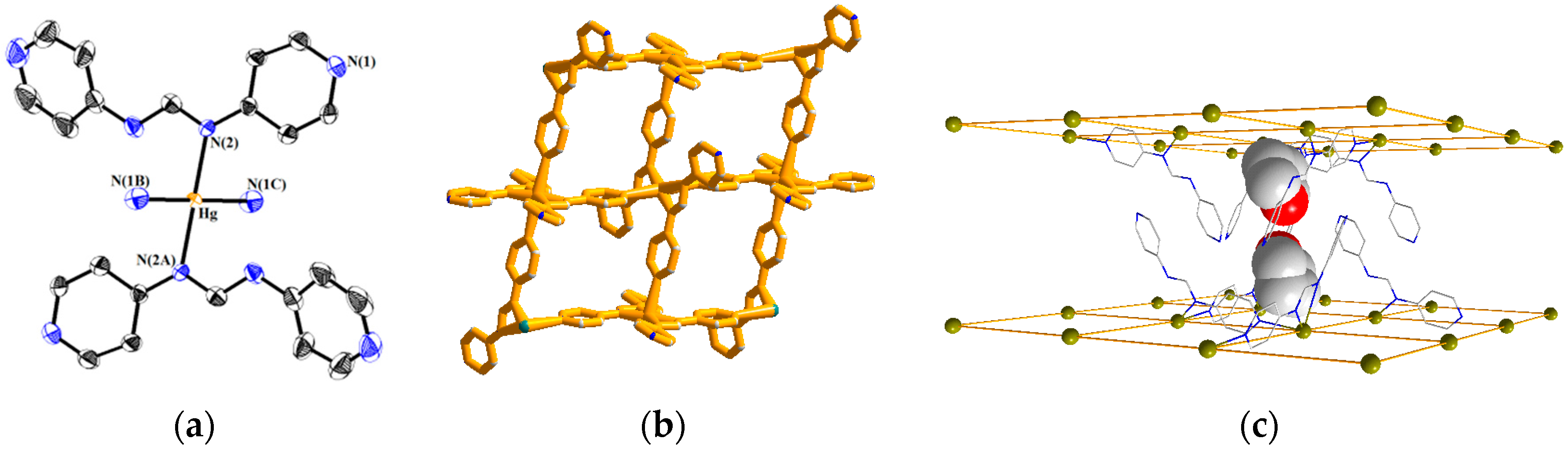
3.2. Structure of 1
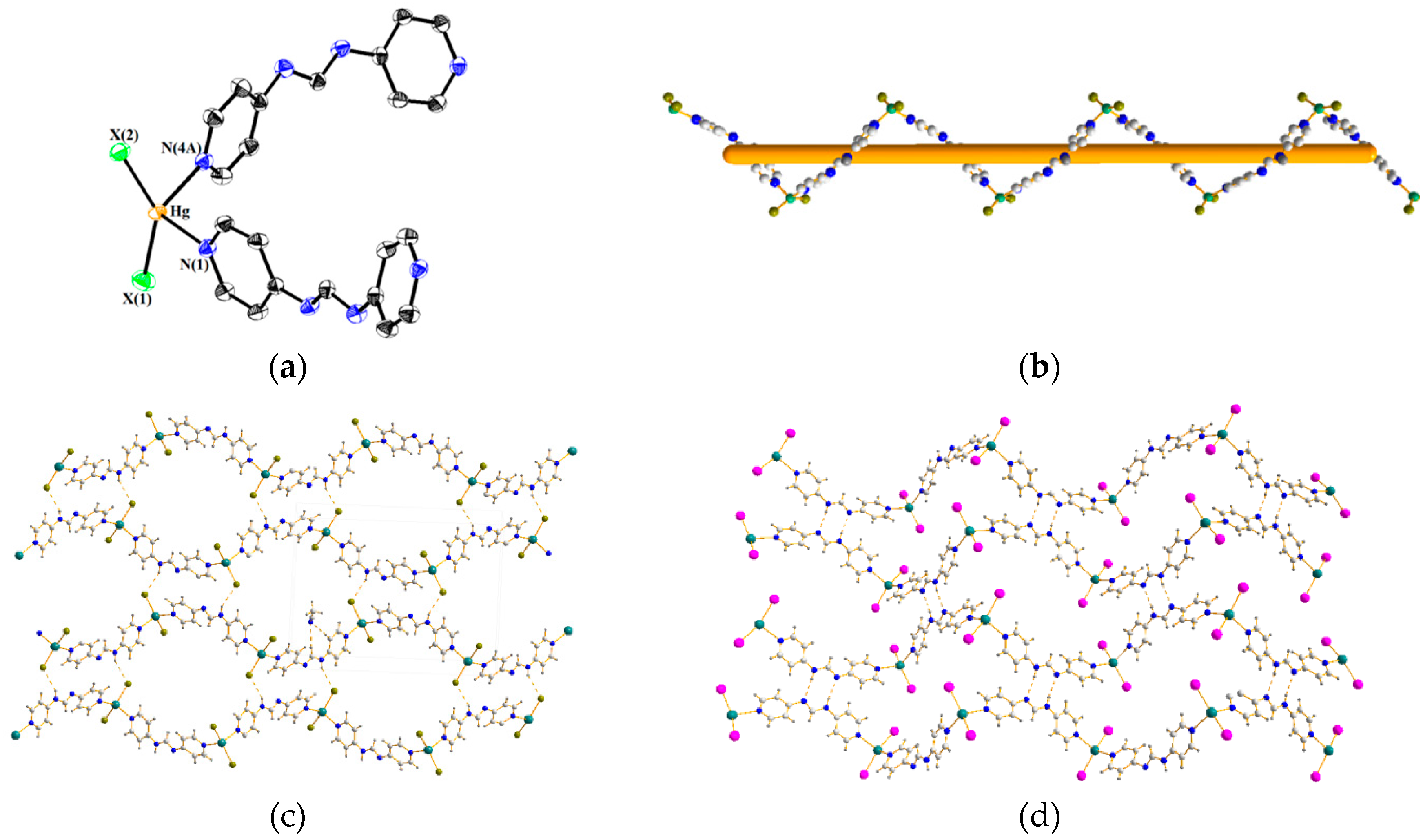
3.3. Structures of 2 and 3
3.4. PXRD Patterns and Thermal Properties
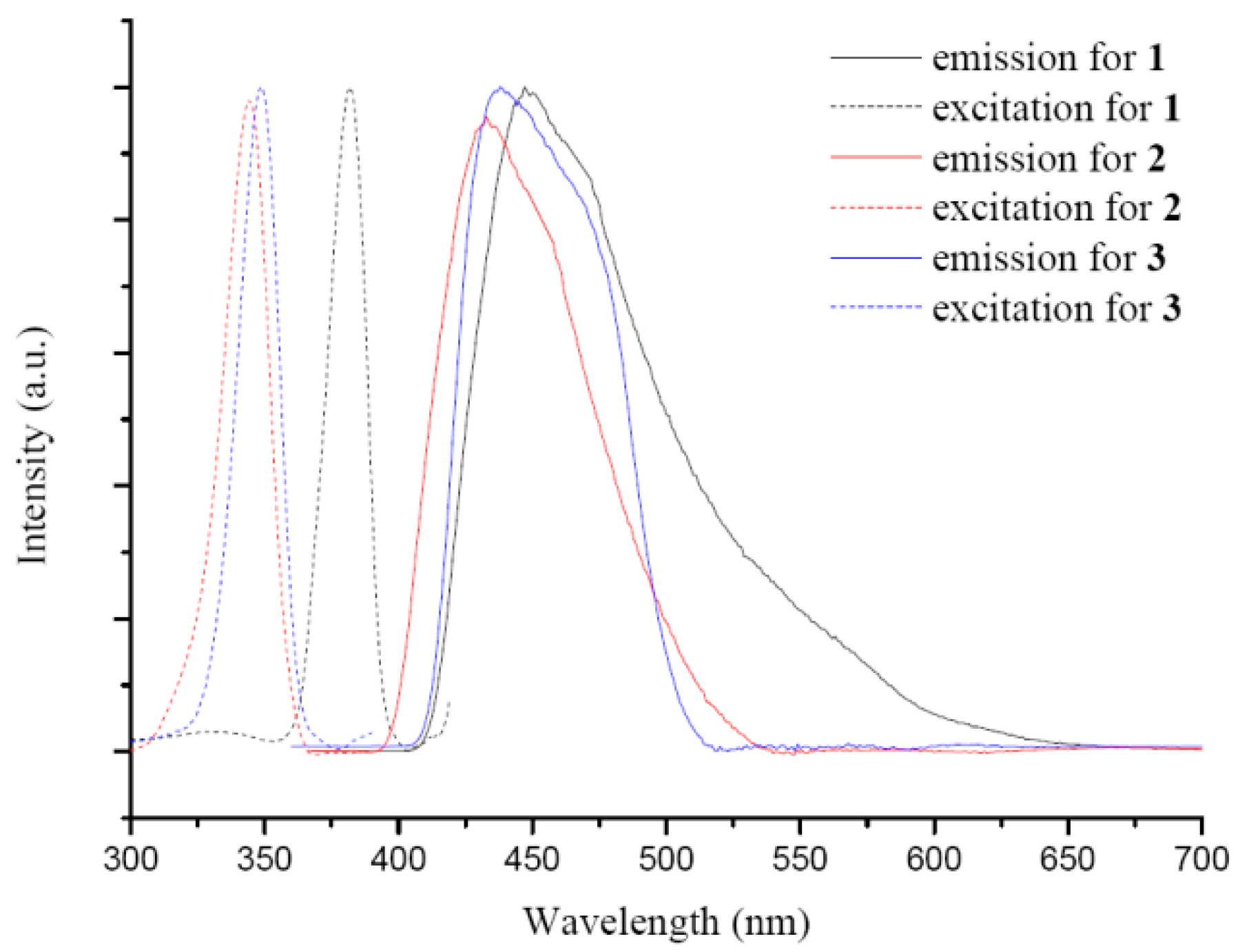
3.5. Luminescent Properties
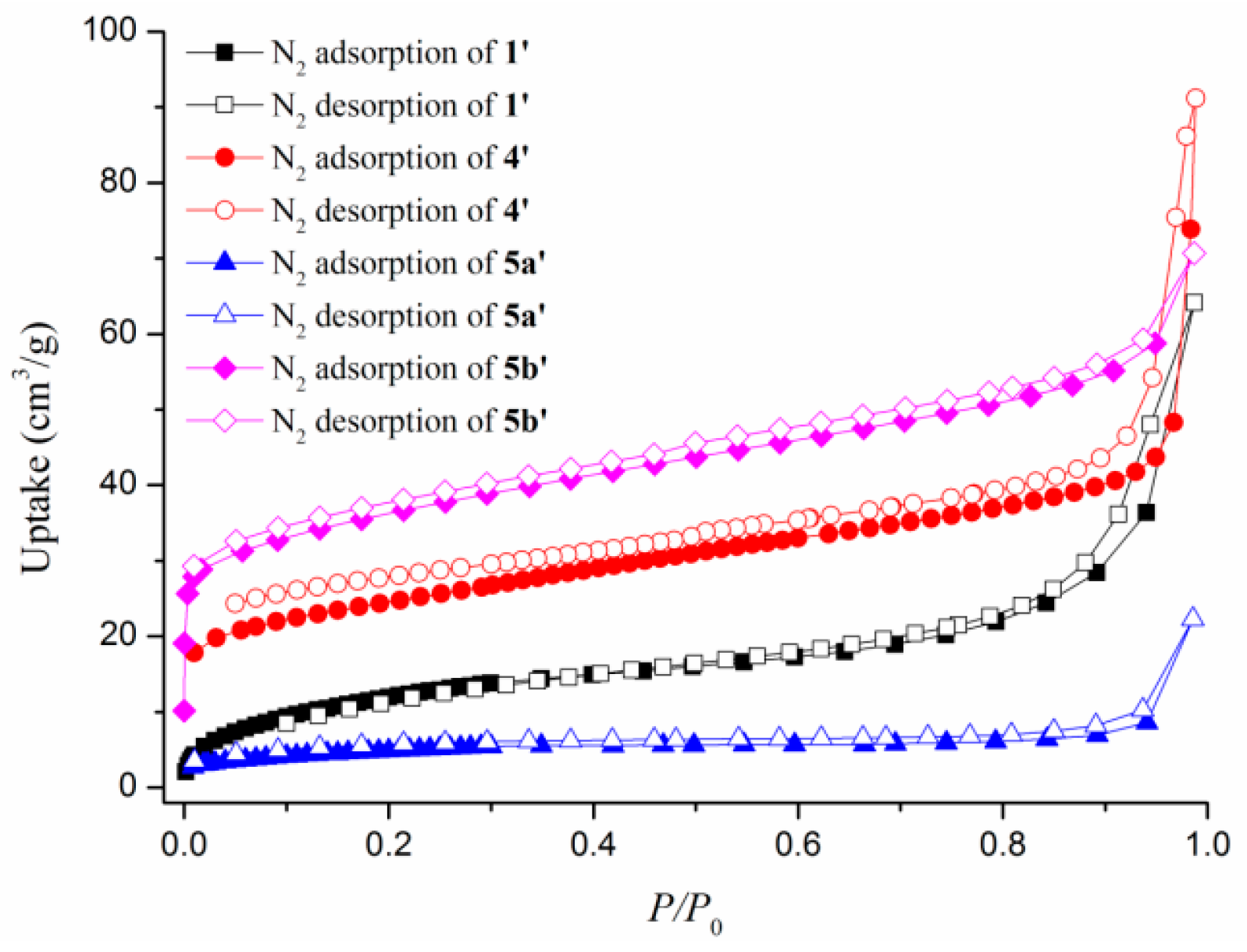
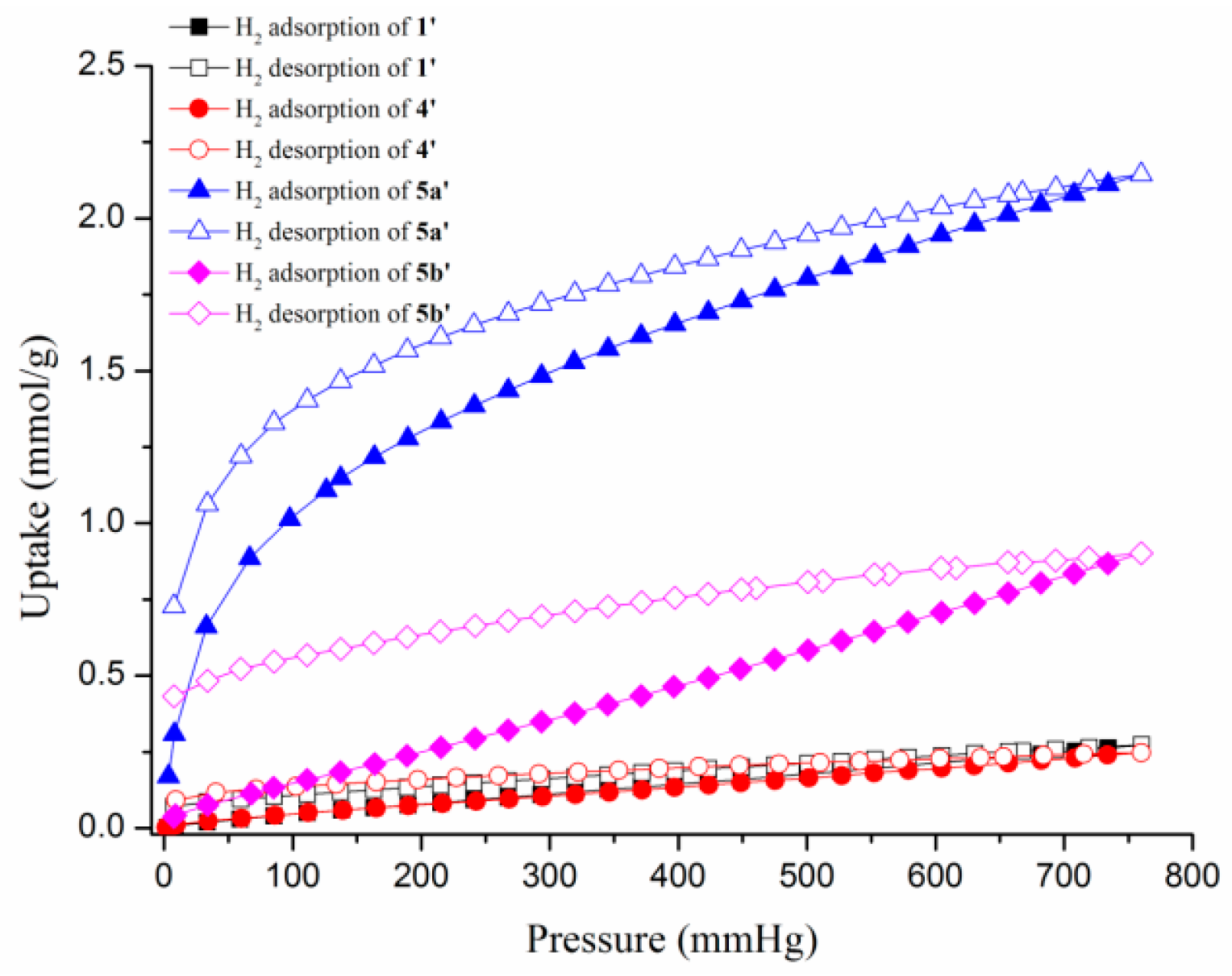
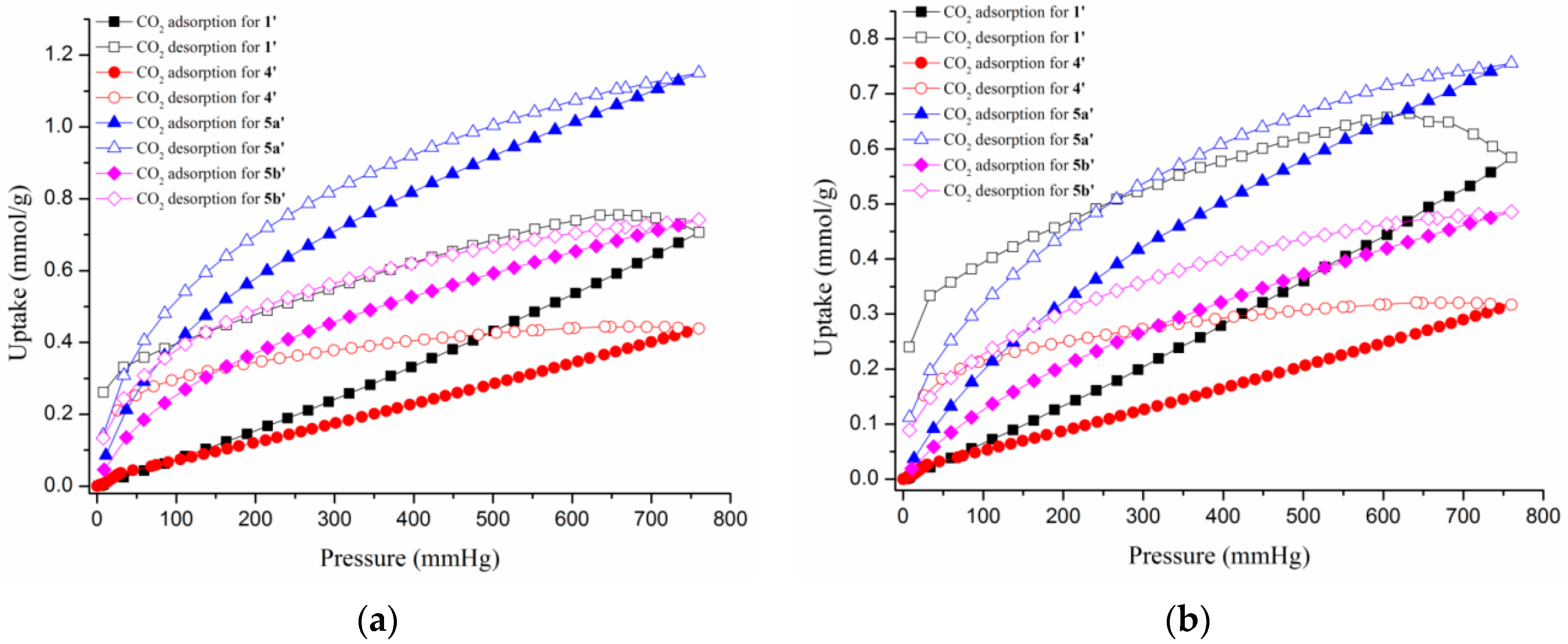
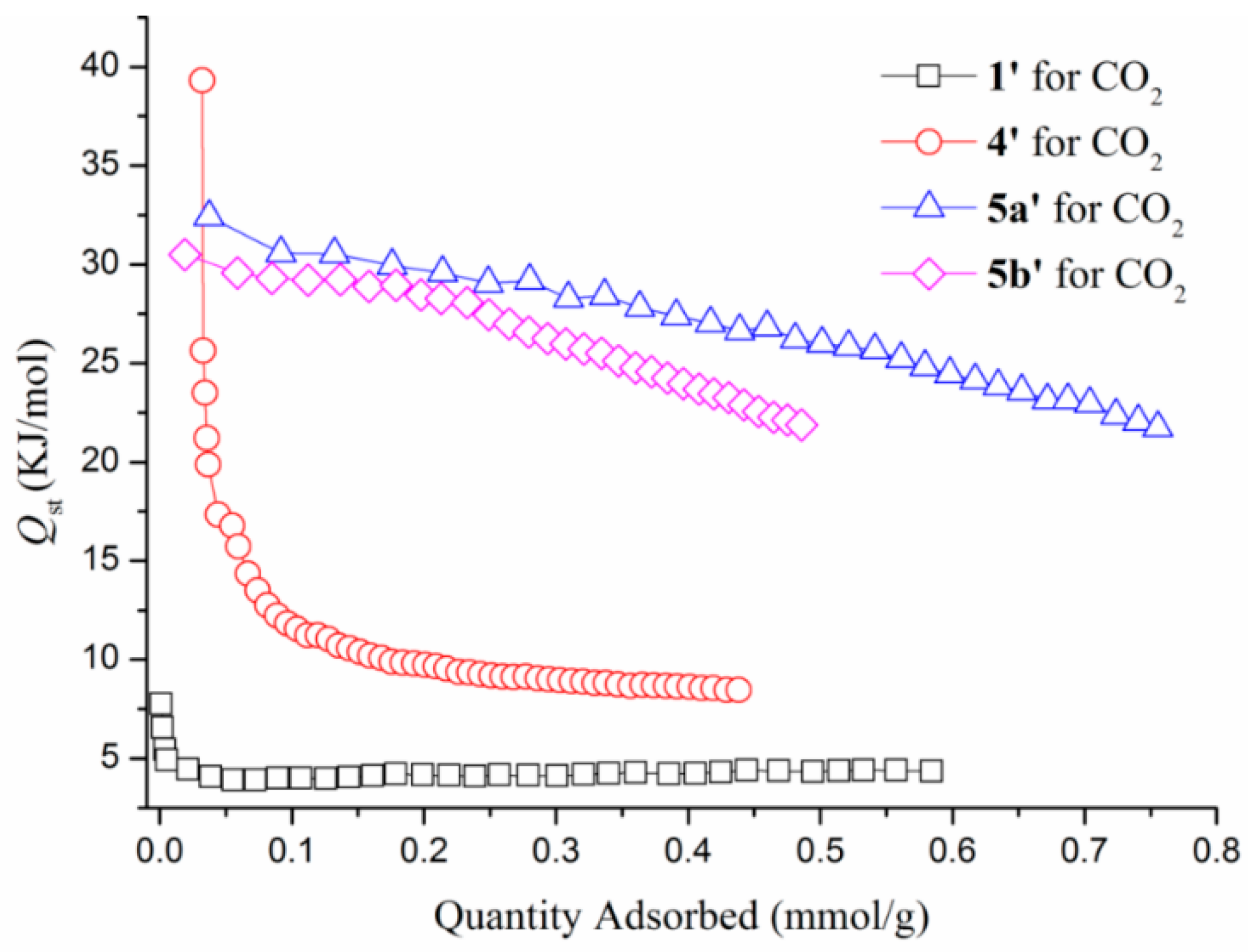
3.6. Gas Sorption Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- O’Keeffe, M.; Yaghi, O.M. Deconstructing the crystal structures of metal-organic frameworks and related materials into their underlying nets. Chem. Rev. 2012, 112, 675–702. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Noro, S.; Nakamura, T. Pore surface engineering of microporous coordination polymers. Chem. Comm. 2006, 7, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Li, J.R.; Sculley, J.; Zhou, H.C. Metal-organic frameworks for separations. Chem. Rev. 2012, 112, 869–932. [Google Scholar] [CrossRef] [PubMed]
- Tiekink, E.R.T.; Vittal, J.J. Frontiers in Crystal Engineering; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Leong, W.L.; Vittal, J.J. One-dimensional coordination polymers: Complexity and diversity in structures, properties and application. Chem. Rev. 2011, 111, 688–764. [Google Scholar] [CrossRef] [PubMed]
- Gable, R.W.; Hoskins, B.F.; Robson, R. A new type of interpenetration involving enmeshed independent square grid sheets. The structure of diaquabis-(4,4′-bipyridine)zinc. J. Chem. Soc. Chem. Commun. 1990, 23, 1677. [Google Scholar] [CrossRef]
- Batten, S.R.; Neville, S.M.; Turner, D.R. Coordination Polymers: Design, Analysis and Application; Royal Society of Chemistry: Cambridge, UK, 2009. [Google Scholar]
- Ye, B.H.; Tong, M.L.; Chen, X.M. Metal-organic molecular architechtures with 2,2’-bipyridl-like and carboxylate ligands. Coord. Chem. Rev. 2005, 249, 545–565. [Google Scholar] [CrossRef]
- Robson, R.; Abrahams, B.E.; Batten, S.R.; Gable, R.W.; Hoskins, B.F.; Lieu, J. Supramolecular Architecture; ACS publications: Washington, DC, USA, 1992. [Google Scholar]
- Cotton, F.A.; Murillo, C.A.; Walton, R.A. Multiple Bonds between Metal Atoms, 3rd ed.; Springer Science and Business Media Inc.: New York, NY, USA, 2005. [Google Scholar]
- Chan, Z.K.; Xu, Y.Y.; Chen, J.D.; Yeh, C.Y.; Wang, C.C.; Tsai, Y.F.; Wang, J.C. Linear and cyclic tetranuclear copper(I) complexes containing anions of N,N’-bis(pyrimidine-2-yl)formamidine. Dalton Trans. 2005, 5, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Chan, Z.K.; Lin, C.H.; Wang, C.C.; Chen, J.D.; Wang, J.C.; Liu, C.W. Syntheses, structures and magnetism of linear tri- and tetra-copper chains containing anions of N,N’-bis(pyrimidine-2-yl)formamidine. Dalton Trans. 2008, 16, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Chan, Z.K.; Chen, T.R.; Chen, J.D.; Wang, J.C.; Liu, C.W. Crystal structures and solution behaviors of dinuclear d10-metal complexes containing anions of N,N’-bis(pyrimidine-2-yl)formamidine. Dalton Trans. 2007, 31, 3450–3458. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.; Li, Y.S.; He, H.Y.; Chen, K.T.; Wu, H.S.; Proserpio, D.M.; Chen, J.D.; Wang, J.C. Stepwise formation of heteronuclear coordination networks based on quadruple-bonded dimolybdenum units containing formamidinate ligands. CrystEngComm 2014, 16, 7385–7388. [Google Scholar] [CrossRef]
- Hsu, W.; Chen, K.T.; Li, Y.S.; Cheng, P.W.; Chen, T.R.; Chen, J.D. Crystal-to-crystal transformations and photoluminescence changes in the Cu(I) coordination networks based on a formamidinate ligand. CrystEngComm 2014, 16, 10640–10648. [Google Scholar] [CrossRef]
- Bruker AXS Inc. Bruker AXS, APEX2, V2008.6; SADABS V2008/1; SAINT V7.60A; SHELXTL V6.14; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Riley, K.E.; Merz, K.M., Jr. Assessment of density functional theory methods for the computation of heats of formation and ionization potentials of systems containing third row transition metals. J. Phys. Chem. A 2007, 111, 6044–6053. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision B.01; Gaussian, Inc.: Wallingford, UK, 2010. [Google Scholar]
- Petitjean, A.; Xing, L.; Wang, R. Functional formamidines: Pyridine substituents make an exception in the usual doubly hydrogen-bonded formamidine dimer. CrystEngComm 2010, 12, 1397–1400. [Google Scholar] [CrossRef]
- Blatov, A.; Shevchenko, A.P.; Proserpio, D.M. Applied topological analysis of crystal structures with the program package ToposPro. Cryst. Growth Des. 2014, 14, 3576–3586. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.M.; Shen, D.; Bülow, M.; Lau, M.L.; Deng, S.; Fitch, F.R.; Lemcoff, N.O.; Semanscin, L. Metallo-organic molecular sieve for gas separation and purification. Microporous Mesoporous Mater. 2002, 55, 217–230. [Google Scholar] [CrossRef]
- Dunne, J.A.; Rao, M.; Sircar, S.; Gorte, R.J.; Myers, A.L. Calorimetric heats of adsorption and adsorption isotherms. 2. O2, N2, Ar, CO2, CH4, C2H6, and SF6 on NaX, H-ZSM-5, and Na-ZSM-5 Zeolites. Langmuir 1996, 12, 5896–5904. [Google Scholar] [CrossRef]
- Nugent, P.; Belmabkhout, Y.; Burd, S.D.; Cairns, A.J.; Luebke, R.; Forrest, K.; Pham, T.; Ma, S.; Space, B.; Wojtas, L.; et al. Porous materials with optimal adsorption thermodynamics and kinetics for CO2 separation. Nature 2013, 495, 80–84. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 4-Hpyf·0.16THF | 1 | 2 | 3 | |
|---|---|---|---|---|
| Formula | C11.67H11.33N4O0.17 | C30H34HgN8O2 | C13H13Br2HgN5 | C13H13HgI2N5 |
| fw | 210.25 | 739.24 | 599.69 | 693.67 |
| Space group | Pī | P21/c | P21/c | P21/c |
| a, Å | 10.7902(1) | 11.3357(3) | 4.9204(1) | 5.1343(2) |
| b, Å | 16.2328(2) | 12.7290(3) | 21.3903(4) | 21.1146(9) |
| c, Å | 19.7864(2) | 10.5421(2) | 16.2077(3) | 16.6380(8) |
| α, deg | 101.218(1) | 90 | 90 | 90 |
| β, deg | 93.271(1) | 91.331(1) | 92.653(1) | 96.902(2) |
| γ, deg | 106.720(1) | 90 | 90 | 90 |
| V, Å3 | 3,232.10(6) | 1,520.73(6) | 1,704.01(6) | 1,790.63(13) |
| Z | 12 | 2 | 4 | 4 |
| Dcalc, g·cm−3 | 1.296 | 1.614 | 2.338 | 2.573 |
| μ, mm−1 | 0.084 | 5.101 | 13.722 | 12.044 |
| No. of reflns meased | 51,996 | 13,961 | 17,060 | 13,570 |
| Independent reflections Rint | 12,722 (0.0331) | 2,977 (0.0400) | 3,321 (0.0518) | 3,526 (0.0650) |
| No. of params | 856 | 182 | 194 | 190 |
| Quality-of-fit indicator c | 1.040 | 1.087 | 1.053 | 1.069 |
| Final R indices [I > 2σ(I)] a,b | R1 = 0.0578, wR2 = 0.1478 | R1 = 0.0537, wR2 = 0.1788 | R1 = 0.0363, wR2 = 0.1014 | R1 = 0.0504, wR2 = 0.1246 |
| R indices | R1 = 0.1142 | R1 = 0.0651 | R1 = 0.0482 | R1 = 0.0634 |
| (All data) | wR2 = 0.1789 | wR2 = 0.1971 | wR2 = 0.1077 | wR2 = 0.1313 |
| 1 | |||
| Hg–N(2) | 2.075(5) | N(2)–Hg–N(2A) | 180.0(2) |
| Hg–N(2A) | 2.075(5) | N(2)–Hg–N(1B) | 94.3(2) |
| Hg–N(1B) | 2.690(6) | N(2A)–Hg–N(1B) | 85.7(2) |
| Hg–N(1C) | 2.690(6) | N(2)–Hg–N(1C) | 85.7(2) |
| N(2A)–Hg–N(1C) | 94.3(2) | ||
| N(1B)–Hg–N(1C) | 180.0 | ||
| 2 | |||
| Hg–N(4A) | 2.362(5) | N(4A)–Hg–N(1) | 108.04(19) |
| Hg–N(1) | 2.366(5) | N(4A)–Hg–Br(1) | 102.77(14) |
| Hg–Br(1) | 2.4923(8) | N(4A)–Hg–Br(2) | 99.27(13) |
| Hg–Br(2) | 2.5425(8) | N(1)–Hg–Br(1) | 100.26(13) |
| N(1)–Hg–Br(2) | 97.16(13) | ||
| Br(1)–Hg–Br(2) | 145.94(3) | ||
| 3 | |||
| Hg–N(4A) | 2.450(7) | N(4A)–Hg–N(1) | 105.6(3) |
| Hg–N(1) | 2.439(8) | N(4A)–Hg–I(1) | 97.9(2) |
| Hg–I(1) | 2.6541(8) | N(4A)–Hg–I(2) | 102.0(2) |
| Hg–I(2) | 2.6415(8) | N(1)–Hg–I(1) | 99.58(18) |
| N(1)–Hg–I(2) | 98.41(18) | ||
| I(1)–Hg–I(2) | 148.40(3) | ||
| 1 | 2 | 3 | |
|---|---|---|---|
| LUMO+2 | −1.92 | −1.85 | −1.85 |
| LUMO+1 | −2.28 | −2.79 | −2.61 |
| LUMO | −2.54 | −2.82 | −2.64 |
| HOMO | −6.17 | −6.66 | −6.44 |
| HOMO−1 | −6.26 | −6.66 | −6.44 |
| HOMO−2 | −6.63 | −6.68 | −6.45 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, W.; Yang, X.-K.; Chhetri, P.M.; Chen, J.-D. Hg(II) Coordination Polymers Based on N,N’-bis(pyridine-4-yl)formamidine. Polymers 2016, 8, 137. https://doi.org/10.3390/polym8040137
Hsu W, Yang X-K, Chhetri PM, Chen J-D. Hg(II) Coordination Polymers Based on N,N’-bis(pyridine-4-yl)formamidine. Polymers. 2016; 8(4):137. https://doi.org/10.3390/polym8040137
Chicago/Turabian StyleHsu, Wayne, Xiang-Kai Yang, Pradhumna Mahat Chhetri, and Jhy-Der Chen. 2016. "Hg(II) Coordination Polymers Based on N,N’-bis(pyridine-4-yl)formamidine" Polymers 8, no. 4: 137. https://doi.org/10.3390/polym8040137
APA StyleHsu, W., Yang, X.-K., Chhetri, P. M., & Chen, J.-D. (2016). Hg(II) Coordination Polymers Based on N,N’-bis(pyridine-4-yl)formamidine. Polymers, 8(4), 137. https://doi.org/10.3390/polym8040137