
The SC-35 Splicing Factor Interacts with RNA Pol II and A-Type Lamin Depletion Weakens This Interaction

 , ,
, ,  ,
, 
Abstract
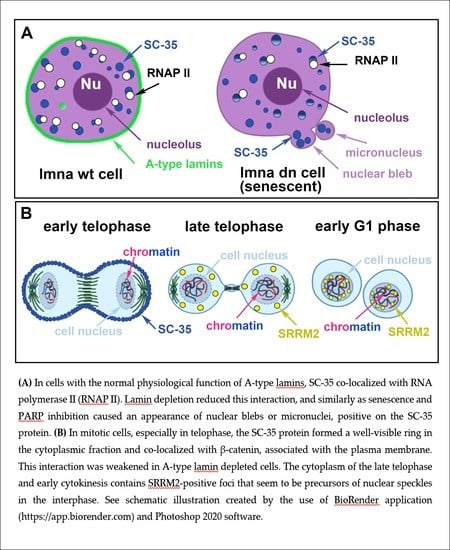
:
1. Introduction
2. Materials and Methods
2.1. Cell Cultivation and Treatment
2.2. Immunofluorescence Staining
2.3. Western Blot
2.4. Laser Scanning Confocal Microscopy
2.5. FLIM-FRET Technique
2.6. FRAP Analysis
2.7. Statistical Analysis
3. Results
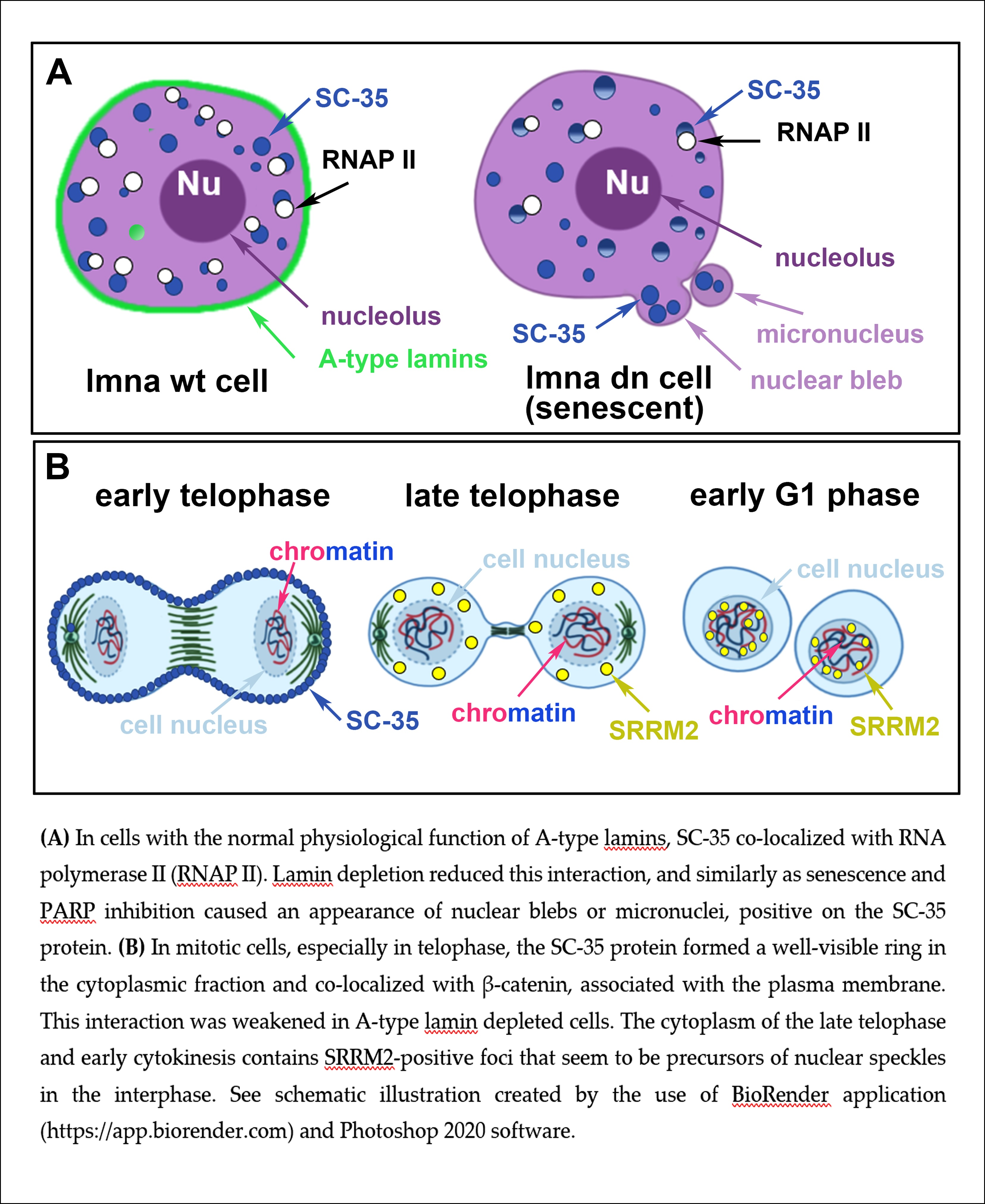
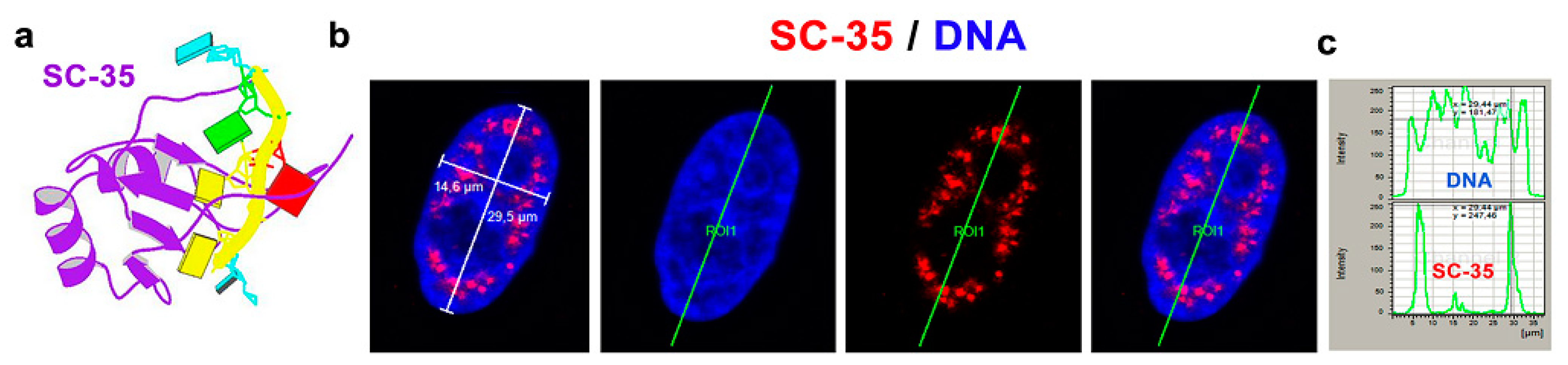
3.1. The SC-35 Protein Was Localized in Nuclear Blebs, and Inhibitors of RNA Polymerases Changed Its Level
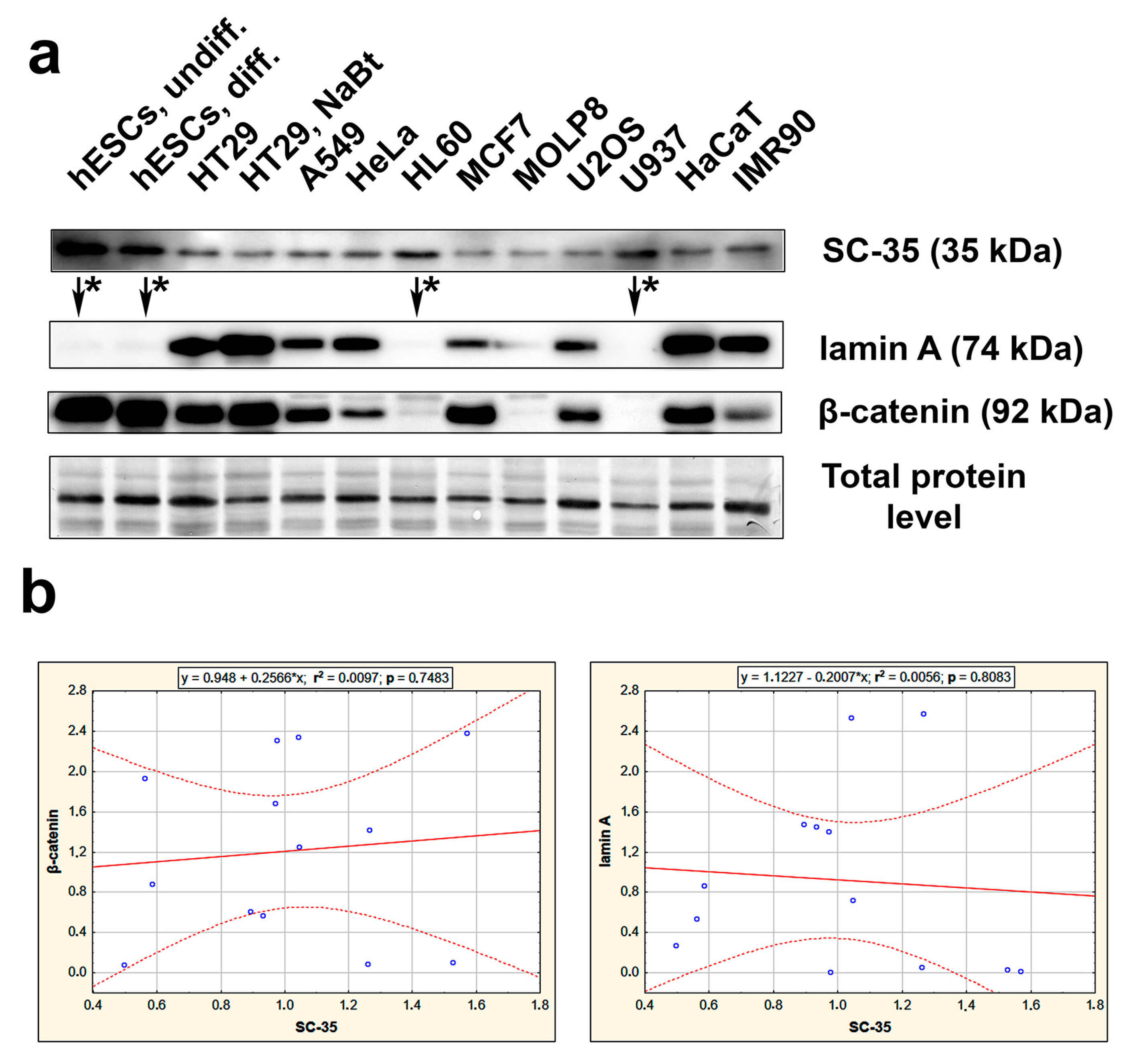
3.2. The Highest Level of SC-35 Was Accompanied by Depletion of A-Type Lamins in Distinct Cell Types
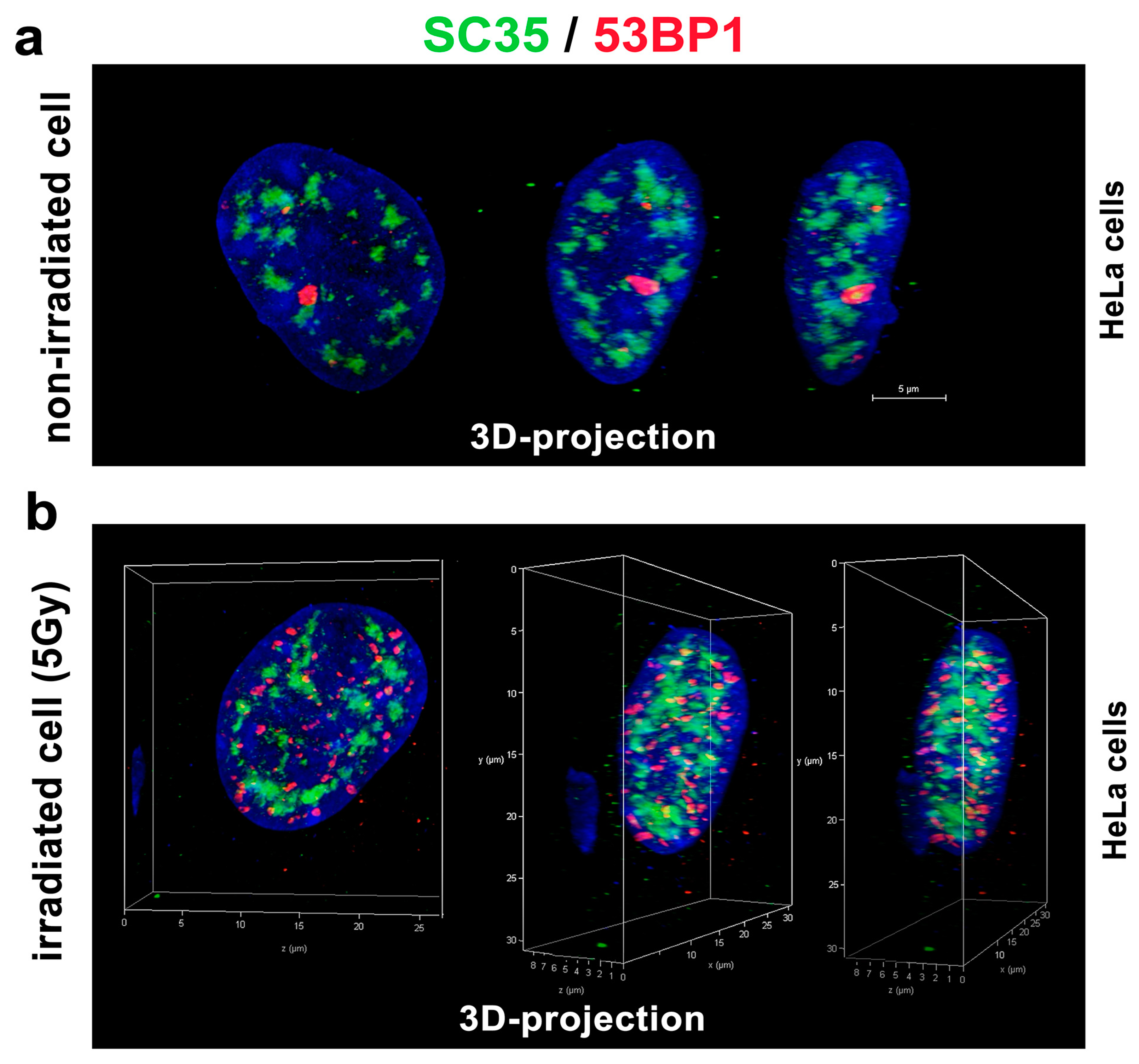
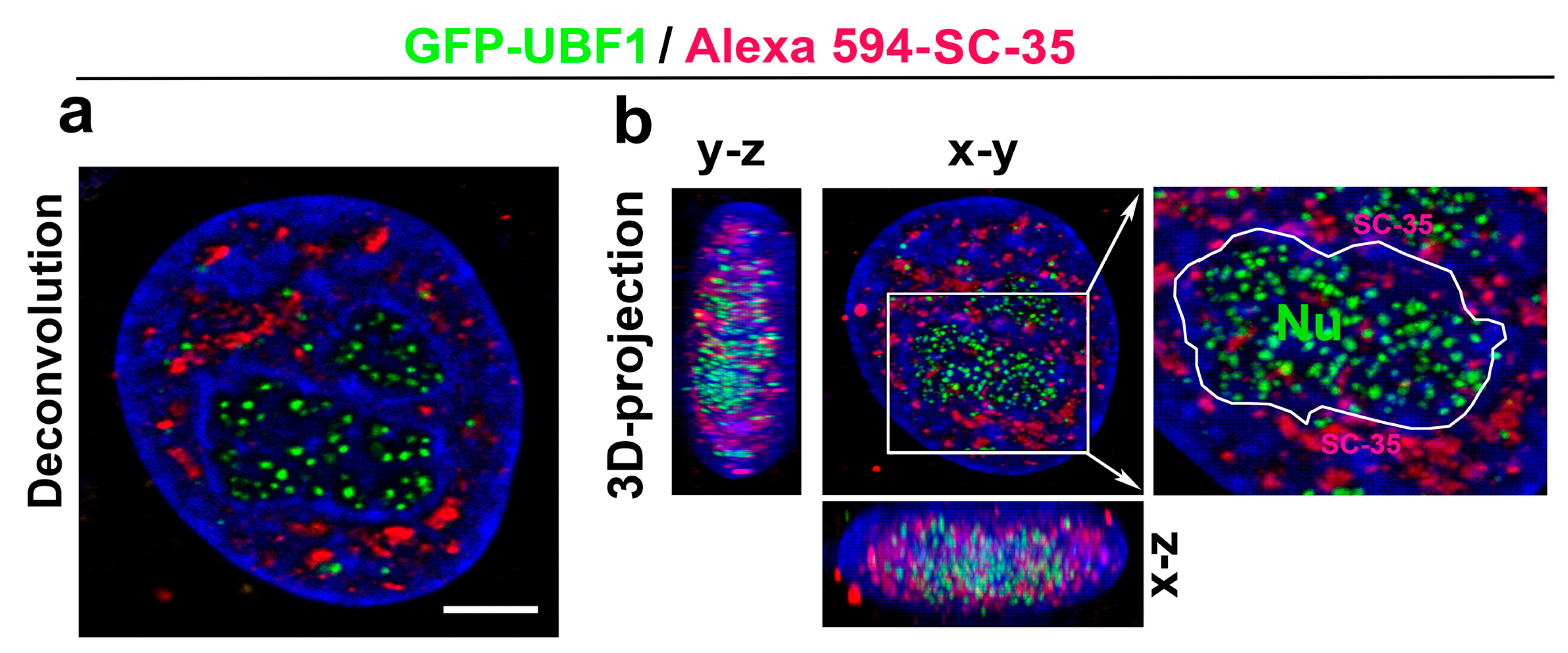
3.3. A Spatial Link of SC-35 Positive Nuclear Speckles to DNA Repair Proteins and Nucleoli
3.4. The SC-35 Protein Decorates the Plasma Membrane in Mitotic Cells and the Degree of Its Colocalization with PCNA Is Enhanced in the Late S-phase of the Cell Cycle
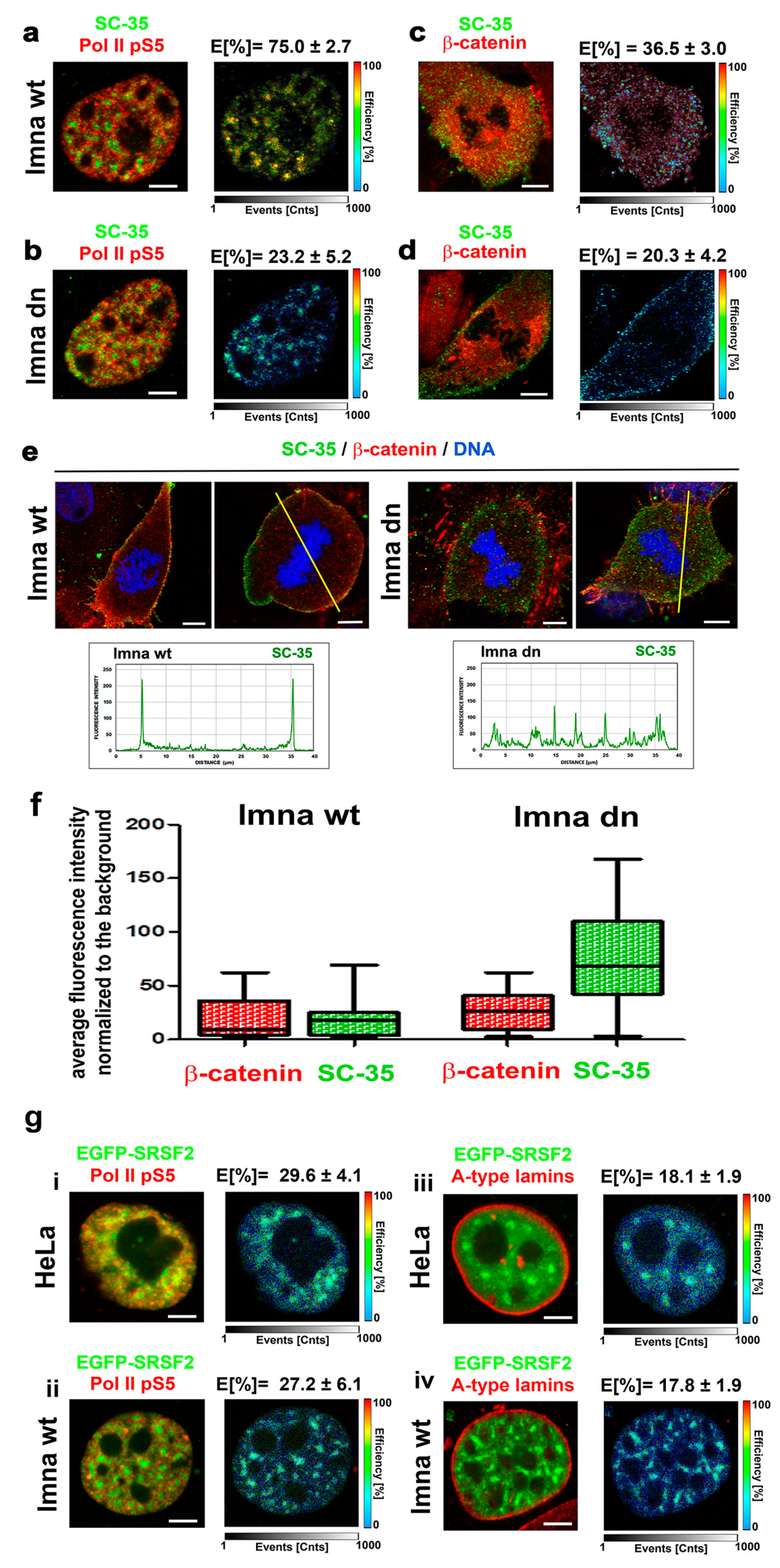
3.5. A-Type Lamin-Dependent Interaction between the SC-35 Protein and RNA Polymerase II in Interphase Cells, or SC-35 and β-catenin Interaction in Mitosis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Misteli, T.; Spector, D.L. RNA polymerase II targets pre-mRNA splicing factors to transcription sites in vivo. Mol. Cell 1999, 3, 697–705. [Google Scholar] [CrossRef]
- Misteli, T.; Caceres, J.F.; Spector, D.L. The dynamics of a pre-mRNA splicing factor in living cells. Nature 1997, 387, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Fakan, S. Perichromatin fibrils are in situ forms of nascent transcripts. Trends Cell Biol. 1994, 4, 86–90. [Google Scholar] [CrossRef]
- Lamond, A.I.; Spector, D.L. Nuclear speckles: A model for nuclear organelles. Nat. Rev. Mol. Cell Biol. 2003, 4, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Spector, D.L. Nuclear organization of pre-mRNA processing. Curr. Opin. Cell Biol. 1993, 5, 442–447. [Google Scholar] [CrossRef]
- Prasanth, K.V.; Sacco-Bubulya, P.A.; Prasanth, S.G.; Spector, D.L. Sequential entry of components of the gene expression machinery into daughter nuclei. Mol. Biol. Cell 2003, 14, 1043–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, K.; Parnaik, V.K. Differential dynamics of splicing factor SC35 during the cell cycle. J. Biosci. 2008, 33, 345–354. [Google Scholar] [CrossRef]
- Dorn, R.; Reuter, G.; Loewendorf, A. Transgene analysis proves mRNA trans-splicing at the complex mod(mdg4) locus in Drosophila. Proc. Natl. Acad. Sci. USA 2001, 98, 9724–9729. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, T.; Giniger, E.; Aigaki, T. Alternative trans-splicing of constant and variable exons of a Drosophila axon guidance gene, lola. Genes Dev. 2003, 17, 2496–2501. [Google Scholar] [CrossRef] [Green Version]
- McManus, C.J.; Duff, M.O.; Eipper-Mains, J.; Graveley, B.R. Global analysis of trans-splicing in Drosophila. Proc. Natl. Acad. Sci. USA 2010, 107, 12975–12979. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, J.; Ma, X.; Sklar, J. Gene fusions and RNA trans-splicing in normal and neoplastic human cells. Cell Cycle 2009, 8, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Heere-Ress, E.; Boucher, B.; Defesche, J.C.; Kastelein, J.; Lavoie, M.A.; Genest, J., Jr. Familial hypercholesterolemia. Acceptor splice site (G-->C) mutation in intron 7 of the LDL-R gene: Alternate RNA editing causes exon 8 skipping or a premature stop codon in exon 8. LDL-R(Honduras-1) [LDL-R1061(-1) G-->C]. Atherosclerosis 1999, 146, 125–131. [Google Scholar] [CrossRef]
- Reid, D.W.; Nicchitta, C.V. Diversity and selectivity in mRNA translation on the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2015, 16, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luco, R.F.; Pan, Q.; Tominaga, K.; Blencowe, B.J.; Pereira-Smith, O.M.; Misteli, T. Regulation of alternative splicing by histone modifications. Science 2010, 327, 996–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.Y.; Zhu, Y.R.; Dai, D.J.; Wang, X.; Jin, H.C. Epigenetic regulation of alternative splicing. Am. J. Cancer Res. 2018, 8, 2346–2358. [Google Scholar]
- Luco, R.F.; Misteli, T. More than a splicing code: Integrating the role of RNA, chromatin and non-coding RNA in alternative splicing regulation. Curr. Opin. Genet. Dev. 2011, 21, 366–372. [Google Scholar] [CrossRef]
- Adhikari, S.; Xiao, W.; Zhao, Y.L.; Yang, Y.G. m(6)A: Signaling for mRNA splicing. RNA Biol. 2016, 13, 756–759. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Patel, B.; Schroeder, K.E.; Raza, A.; Dejong, J. Organization and transcriptional output of a novel mRNA-like piRNA gene (mpiR) located on mouse chromosome 10. RNA 2008, 14, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef]
- Ilik, I.A.; Malszycki, M.; Lubke, A.K.; Schade, C.; Meierhofer, D.; Aktas, T. SON and SRRM2 are essential for nuclear speckle formation. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Rein, I.D.; Landsverk, K.S.; Micci, F.; Patzke, S.; Stokke, T. Replication-induced DNA damage after PARP inhibition causes G2 delay, and cell line-dependent apoptosis, necrosis and multinucleation. Cell Cycle 2015, 14, 3248–3260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Hunt, C.R.; Pandita, R.K.; Kumar, R.; Yang, C.R.; Horikoshi, N.; Bachoo, R.; Serag, S.; Story, M.D.; Shay, J.W.; et al. Lamin A/C depletion enhances DNA damage-induced stalled replication fork arrest. Mol. Cell Biol. 2013, 33, 1210–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartova, E.; Malyskova, B.; Komurkova, D.; Legartova, S.; Suchankova, J.; Krejci, J.; Kozubek, S. Function of heterochromatin protein 1 during DNA repair. Protoplasma 2017, 254, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Essers, J.; Theil, A.F.; Baldeyron, C.; van Cappellen, W.A.; Houtsmuller, A.B.; Kanaar, R.; Vermeulen, W. Nuclear dynamics of PCNA in DNA replication and repair. Mol. Cell Biol. 2005, 25, 9350–9359. [Google Scholar] [CrossRef] [Green Version]
- Stixova, L.; Sehnalova, P.; Legartova, S.; Suchankova, J.; Hruskova, T.; Kozubek, S.; Sorokin, D.V.; Matula, P.; Raska, I.; Kovarik, A.; et al. HP1beta-dependent recruitment of UBF1 to irradiated chromatin occurs simultaneously with CPDs. Epigenetics Chromatin 2014, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- McCrea, P.D.; Turck, C.W.; Gumbiner, B. A homolog of the armadillo protein in Drosophila (plakoglobin) associated with E-cadherin. Science 1991, 254, 1359–1361. [Google Scholar] [CrossRef]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Stixova, L.; Matula, P.; Kozubek, S.; Gombitova, A.; Cmarko, D.; Raska, I.; Bartova, E. Trajectories and nuclear arrangement of PML bodies are influenced by A-type lamin deficiency. Biol. Cell 2012, 104, 418–432. [Google Scholar] [CrossRef]
- Bartova, E.; Pachernik, J.; Harnicarova, A.; Kovarik, A.; Kovarikova, M.; Hofmanova, J.; Skalnikova, M.; Kozubek, M.; Kozubek, S. Nuclear levels and patterns of histone H3 modification and HP1 proteins after inhibition of histone deacetylases. J. Cell Sci. 2005, 118, 5035–5046. [Google Scholar] [CrossRef] [Green Version]
- Bartova, E.; Krejci, J.; Harnicarova, A.; Kozubek, S. Differentiation of human embryonic stem cells induces condensation of chromosome territories and formation of heterochromatin protein 1 foci. Differentiation 2008, 76, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Bartova, E.; Harnicarova, A.; Pachernik, J.; Kozubek, S. Nuclear topography and expression of the BCR/ABL fusion gene and its protein level influenced by cell differentiation and RNA interference. Leuk. Res. 2005, 29, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Lukasova, E.; Kovarˇík, A.; Bac ikova, A.; Falk, M.; Kozubek, S. Loss of lamin B receptor is necessary to induce cellular senescence. Biochem. J. 2017, 474, 281–300. [Google Scholar] [CrossRef] [PubMed]
- Krejci, J.; Harnicarova, A.; Kurova, J.; Uhlirova, R.; Kozubek, S.; Legartova, S.; Hajek, R.; Bartova, E. Nuclear organization of PML bodies in leukaemic and multiple myeloma cells. Leuk. Res. 2008, 32, 1866–1877. [Google Scholar] [CrossRef] [PubMed]
- Ismail, I.H.; Andrin, C.; McDonald, D.; Hendzel, M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 2010, 191, 45–60. [Google Scholar] [CrossRef]
- Svobodova Kovarikova, A.; Stixova, L.; Kovarik, A.; Komurkova, D.; Legartova, S.; Fagherazzi, P.; Bartova, E. N(6)-Adenosine Methylation in RNA and a Reduced m3G/TMG Level in Non-Coding RNAs Appear at Microirradiation-Induced DNA Lesions. Cells 2020, 9, 360. [Google Scholar] [CrossRef] [Green Version]
- Suchankova, J.; Legartova, S.; Ruckova, E.; Vojtesek, B.; Kozubek, S.; Bartova, E. Mutations in the TP53 gene affected recruitment of 53BP1 protein to DNA lesions, but level of 53BP1 was stable after gamma-irradiation that depleted MDC1 protein in specific TP53 mutants. Histochem. Cell Biol. 2017, 148, 239–255. [Google Scholar] [CrossRef]
- Dundr, M.; Hoffmann-Rohrer, U.; Hu, Q.; Grummt, I.; Rothblum, L.I.; Phair, R.D.; Misteli, T. A kinetic framework for a mammalian RNA polymerase in vivo. Science 2002, 298, 1623–1626. [Google Scholar] [CrossRef]
- Bartova, E.; Sustackova, G.; Stixova, L.; Kozubek, S.; Legartova, S.; Foltankova, V. Recruitment of Oct4 protein to UV-damaged chromatin in embryonic stem cells. PLoS ONE 2011, 6, e27281. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, S.; Kim, S.K.; Kubista, M.; Norden, B. Binding of 4’,6-diamidino-2-phenylindole (DAPI) to AT regions of DNA: Evidence for an allosteric conformational change. Biochemistry 1993, 32, 2987–2998. [Google Scholar] [CrossRef]
- Legartova, S.; Jugova, A.; Stixova, L.; Kozubek, S.; Fojtova, M.; Zdrahal, Z.; Lochmanova, G.; Bartova, E. Epigenetic aspects of HP1 exchange kinetics in apoptotic chromatin. Biochimie 2013, 95, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Legartova, S.; Lochmanova, G.; Zdrahal, Z.; Kozubek, S.; Sponer, J.; Krepl, M.; Pokorna, P.; Bartova, E. DNA Damage Changes Distribution Pattern and Levels of HP1 Protein Isoforms in the Nucleolus and Increases Phosphorylation of HP1beta-Ser88. Cells 2019, 8, 1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmond, V.; Moysan, E.; Khochbin, S.; Matthias, P.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. Acetylation and phosphorylation of SRSF2 control cell fate decision in response to cisplatin. EMBO J. 2011, 30, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Alibhai, D.; Margineanu, A.; Laine, R.; Kennedy, G.; McGinty, J.; Warren, S.; Kelly, D.; Alexandrov, Y.; Munro, I.; et al. FLIM FRET technology for drug discovery: Automated multiwell-plate high-content analysis, multiplexed readouts and application in situ. Chemphyschem 2011, 12, 609–626. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R.; Gryczynski, I.I.; Gryczynski, Z. High Throughput Screening with Multiphoton Excitation. J. Biomol. Screen 1999, 4, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sillen, A.; Engelborghs, Y. The correct use of “average” fluorescence parameters. Photochem. Photobiol. 1998, 67, 475–486. [Google Scholar] [CrossRef]
- Daubner, G.M.; Clery, A.; Jayne, S.; Stevenin, J.; Allain, F.H. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 2012, 31, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Legartova, S.; Sehnalova, P.; Malyskova, B.; Kuntziger, T.; Collas, P.; Cmarko, D.; Raska, I.; Sorokin, D.V.; Kozubek, S.; Bartova, E. Localized Movement and Levels of 53BP1 Protein Are Changed by gamma-irradiation in PML Deficient Cells. J. Cell Biochem. 2016, 117, 2583–2596. [Google Scholar] [CrossRef] [PubMed]
- Bubulya, P.A.; Prasanth, K.V.; Deerinck, T.J.; Gerlich, D.; Beaudouin, J.; Ellisman, M.H.; Ellenberg, J.; Spector, D.L. Hypophosphorylated SR splicing factors transiently localize around active nucleolar organizing regions in telophase daughter nuclei. J. Cell Biol. 2004, 167, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Spector, D.L.; Lamond, A.I. Nuclear speckles. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [Green Version]
- Galganski, L.; Urbanek, M.O.; Krzyzosiak, W.J. Nuclear speckles: Molecular organization, biological function and role in disease. Nucleic Acids Res. 2017, 45, 10350–10368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.D.; Maniatis, T. The 35-kDa mammalian splicing factor SC35 mediates specific interactions between U1 and U2 small nuclear ribonucleoprotein particles at the 3′ splice site. Proc. Natl. Acad. Sci. USA 1992, 89, 1725–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neugebauer, K.M. On the importance of being co-transcriptional. J. Cell Sci. 2002, 115, 3865–3871. [Google Scholar] [CrossRef] [Green Version]
- Shimi, T.; Pfleghaar, K.; Kojima, S.; Pack, C.G.; Solovei, I.; Goldman, A.E.; Adam, S.A.; Shumaker, D.K.; Kinjo, M.; Cremer, T.; et al. The A- and B-type nuclear lamin networks: Microdomains involved in chromatin organization and transcription. Genes Dev. 2008, 22, 3409–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bercht Pfleghaar, K.; Taimen, P.; Butin-Israeli, V.; Shimi, T.; Langer-Freitag, S.; Markaki, Y.; Goldman, A.E.; Wehnert, M.; Goldman, R.D. Gene-rich chromosomal regions are preferentially localized in the lamin B deficient nuclear blebs of atypical progeria cells. Nucleus 2015, 6, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funkhouser, C.M.; Sknepnek, R.; Shimi, T.; Goldman, A.E.; Goldman, R.D.; Olvera de la Cruz, M. Mechanical model of blebbing in nuclear lamin meshworks. Proc. Natl. Acad. Sci. USA 2013, 110, 3248–3253. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Suarez, I.; Redwood, A.B.; Gonzalo, S. Loss of A-type lamins and genomic instability. Cell Cycle 2009, 8, 3860–3865. [Google Scholar] [CrossRef]
- Caruso, R.A.; Fedele, F.; Crisafulli, C.; Paparo, D.; Parisi, A.; Luciano, R.; Cavallari, V. Abnormal nuclear structures (micronuclei, nuclear blebs, strings, and pockets) in a case of anaplastic giant cell carcinoma of the thyroid: An immunohistochemical and ultrastructural study. Ultrastruct. Pathol. 2011, 35, 14–18. [Google Scholar] [CrossRef]
- Utani, K.; Okamoto, A.; Shimizu, N. Generations of micronuclei during interphase by coupling between cytoplasmic membrane blebbing and nuclear budding. PLoS ONE 2011, 6, e27233. [Google Scholar] [CrossRef] [Green Version]
- Rai, A.K.; Chen, J.X.; Selbach, M.; Pelkmans, L. Kinase-controlled phase transition of membraneless organelles in mitosis. Nature 2018, 559, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Dundr, M.; Misteli, T. Biogenesis of nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000711. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Cell Culture Medium | Reference |
|---|---|---|
| A549 (human lung cancer) (#ATCC® CCL-185™, Germany) | DMEM medium supplemented with 10% FCS | [30] |
| HL60 (human acute promyelocytic leukemia) (#ATCC® CCL-240™, Germany) | IMDM medium supplemented with 10% FCS | [32] |
| MCF7 (human adenocarcinoma) (#ATCC® HTB-22™, Germany) | EMEM medium supplemented with 0.01 mg/mL human recombinant insulin and 10% FCS | [33] |
| MOLP8 (human multiple myeloma) (#ACC 569, DSMZ, Germany) | RPMI 1640 medium supplemented with 20% FCS | [34] |
| U2OS (human osteosarcoma) collaboration with the Institute of Biology and Medical Genetics, Charles University in Prague) | DMEM medium supplemented with 10% FCS | [35] |
| U937 (human histiocytic lymphoma) (#ATCC® CRL-1593.2™, Germany) | RPMI 1640 medium supplemented with 10% FCS | [34] |
| HaCaT (human keratinocytes) (#300493, CLS, Germany) | DMEM medium supplemented with 10% FCS | [36] |
| IMR90 (human lung fibroblast) (#ATCC® CCL-186™, Germany) | EMEM medium supplemented with 10% FCS, 1% non-essential amino acids (NEAA), and 2mM glutamine | [37] |
| QYCy3 (DONOR) | ECCy5(M−1cm−1) | QYCy5 (ACCEPTOR) | J(λ) (*1 × 1015 M−1cm−1nm4) | R0(Å) | R0 × QYCy5 |
| 0.15 | 250,000 | 0.30 | 7.6 | 49.73 | 14.92 |
| QYEGFP (DONOR) | EC AF594 * (M−1cm−1) | QYAF594 (ACCEPTOR) | J(λ) (*1 × 1015 M−1cm−1nm4) | R0(Å) | R0 × QYAF594 |
| 0.60 | 92,000 | 0.66 | 1.80 | 49.30 | 32.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Legartová, S.; Fagherazzi, P.; Stixová, L.; Kovařík, A.; Raška, I.; Bártová, E. The SC-35 Splicing Factor Interacts with RNA Pol II and A-Type Lamin Depletion Weakens This Interaction. Cells 2021, 10, 297. https://doi.org/10.3390/cells10020297
Legartová S, Fagherazzi P, Stixová L, Kovařík A, Raška I, Bártová E. The SC-35 Splicing Factor Interacts with RNA Pol II and A-Type Lamin Depletion Weakens This Interaction. Cells. 2021; 10(2):297. https://doi.org/10.3390/cells10020297
Chicago/Turabian StyleLegartová, Soňa, Paolo Fagherazzi, Lenka Stixová, Aleš Kovařík, Ivan Raška, and Eva Bártová. 2021. "The SC-35 Splicing Factor Interacts with RNA Pol II and A-Type Lamin Depletion Weakens This Interaction" Cells 10, no. 2: 297. https://doi.org/10.3390/cells10020297
APA StyleLegartová, S., Fagherazzi, P., Stixová, L., Kovařík, A., Raška, I., & Bártová, E. (2021). The SC-35 Splicing Factor Interacts with RNA Pol II and A-Type Lamin Depletion Weakens This Interaction. Cells, 10(2), 297. https://doi.org/10.3390/cells10020297