
Genetic and Genomic Pathways of Melanoma Development, Invasion and Metastasis


Abstract
: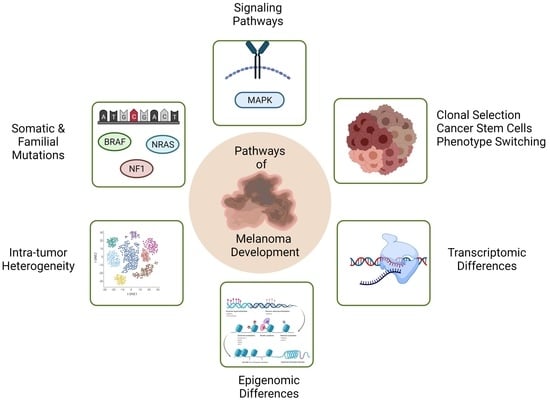
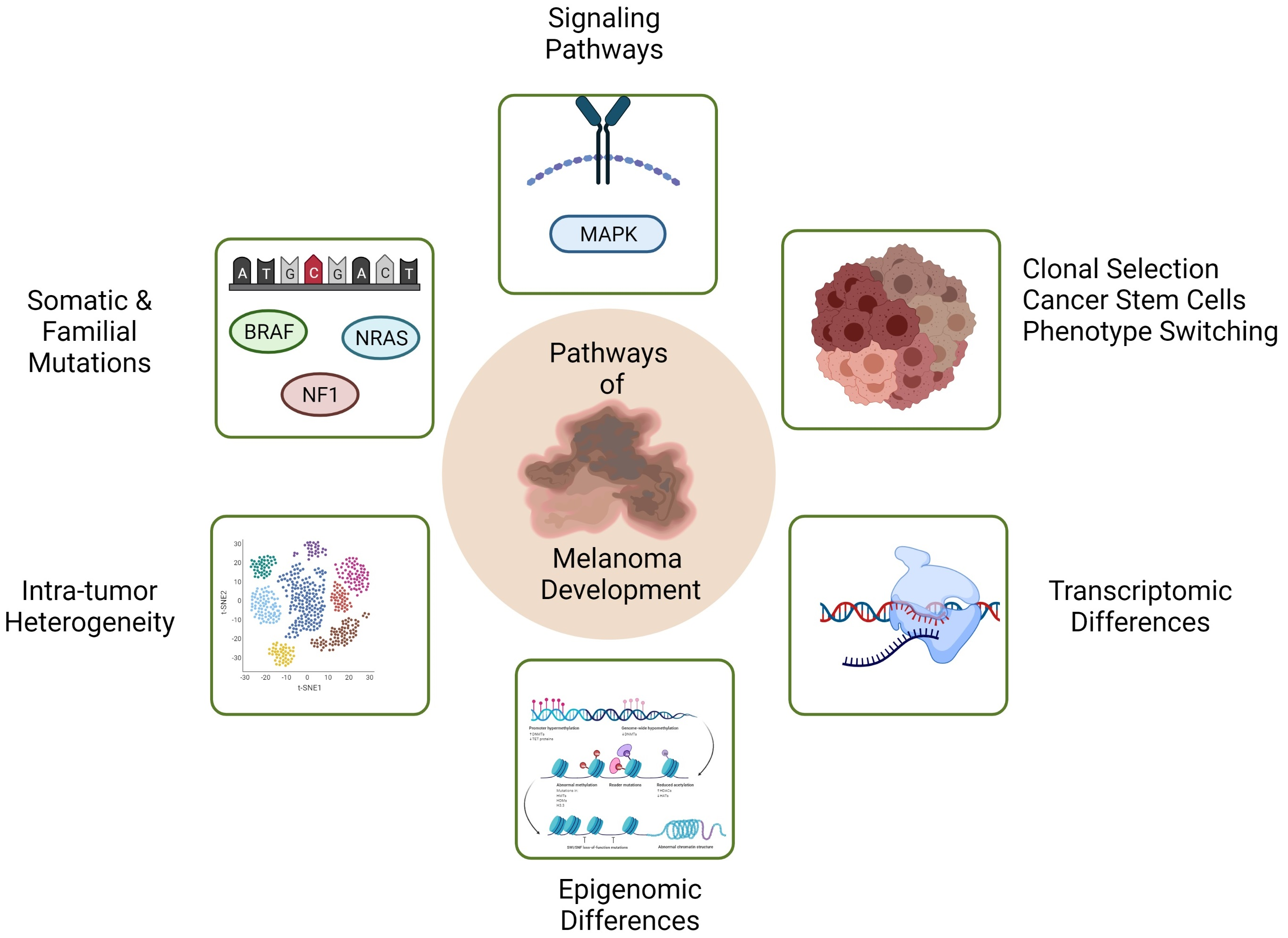{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Mutational Landscapes of Melanoma
2.1. The Mutational Landscape of Somatic Melanomas
2.2. The Mutational Landscape of Familial Melanomas
2.3. Mutational Landscape Studies of Metastatic Melanomas
3. Gene Expression Alterations and Cell Signalling Pathways Underlying Melanoma Initiation and Progression to Invasion and Metastasis
4. Intra-Tumoral Heterogeneity in Melanoma as A Precursor to Metastasis
4.1. The Clonal Evolution Model
4.2. The Cancer Stem Cell Model
4.3. The Phenotype Switching Model
5. Single Cell Sequencing and Spatial Transcriptomics: A Step towards Understanding Tumour Complexity, Tumour Progression and Drug Resistance
6. Transcriptomic or Epigenomic Differences between Invasive and Non-Invasive Phenotypes in Melanoma Cell Lines
6.1. Transcriptomic Differences between Invasive and Non-Invasive Phenotypes
6.2. Epigenomic Differences between Invasive and Non-Invasive Phenotypes
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, A.J.; Mihm, M.C. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Schwartz, G.K.; Tezel, T.; Marr, B.; Francis, J.H.; Nathan, P.D. Metastatic Disease from Uveal Melanoma: Treatment Options and Future Prospects. Br. J. Ophthalmol. 2017, 101, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteman, D.C.; Green, A.C.; Olsen, C.M. The Growing Burden of Invasive Melanoma: Projections of Incidence Rates and Numbers of New Cases in Six Susceptible Populations through 2031. J. Investig. Dermatol. 2016, 136, 1161–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, M.S.; Zager, J.; Faries, M.; Alexander, H.R.; Royal, R.E.; Wood, B.; Choi, J.; McCluskey, K.; Whitman, E.; Agarwala, S.; et al. Results of a Randomized Controlled Multicenter Phase III Trial of Percutaneous Hepatic Perfusion Compared with Best Available Care for Patients with Melanoma Liver Metastases. Ann. Surg. Oncol. 2016, 23, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Liu, W.; Gotlieb, V. The Rapidly Evolving Therapies for Advanced Melanoma—Towards Immunotherapy, Molecular Targeted Therapy, and Beyond. Crit. Rev. Oncol. Hematol. 2016, 99, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.; Fishman, G.A.; Joshi, K.; Jampol, L.M. Chorioretinal Lesions in a Case of Melanoma-Associated Retinopathy Treated With Pembrolizumab. JAMA Ophthalmol. 2016, 134, 1184. [Google Scholar] [CrossRef]
- Queirolo, P.; Pfeffer, U. Metastatic Melanoma: How Research Can Modify the Course of a Disease. Cancer Metastasis Rev. 2017, 36, 3–5. [Google Scholar] [CrossRef]
- Seftor, E.A.; Seftor, R.E.B.; Weldon, D.S.; Kirsammer, G.T.; Margaryan, N.V.; Gilgur, A.; Hendrix, M.J.C. Melanoma Tumor Cell Heterogeneity: A Molecular Approach to Study Subpopulations Expressing the Embryonic Morphogen Nodal. Semin. Oncol. 2014, 41, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Shannan, B.; Perego, M.; Somasundaram, R.; Herlyn, M. Heterogeneity in Melanoma. In Melanoma; Kaufman, H.L., Mehnert, J.M., Eds.; Cancer Treatment and Research; Springer International Publishing: Cham, Switzerland, 2016; Volume 167, pp. 1–15. ISBN 978-3-319-22538-8. [Google Scholar]
- Lawrence, S.K.; Nguyen, D.; Bowen, C.; Richards-Peterson, L.; Skordos, K.W. The Metabolic Drug-Drug Interaction Profile of Dabrafenib: In Vitro Investigations and Quantitative Extrapolation of the P450-Mediated DDI Risk. Drug Metab. Dispos. 2014, 42, 1180–1190. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Wellbrock, C.; Hurlstone, A. BRAF as Therapeutic Target in Melanoma. Biochem. Pharmacol. 2010, 80, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Angelini, S.; Snellman, E.; Hemminki, K. BRAF Mutations Are Common Somatic Events in Melanocytic Nevi. J. Investig. Dermatol. 2004, 122, 342–348. Available online: http://www.blackwellpublishing.com/products/journals/suppmat/jid/jid22225/jid22225sm.htm (accessed on 12 September 2021). [CrossRef] [PubMed] [Green Version]
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS Mutation Frequencies Among Primary Tumors and Metastases in Patients with Melanoma. J. Clin. Oncol. 2012, 30, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, J.L.; Fridlyand, J.; Patel, H.; Jain, A.N.; Busam, K.; Kageshita, T.; Ono, T.; Albertson, D.G.; Pinkel, D.; Bastian, B.C. Determinants of BRAF Mutations in Primary Melanomas. J. Natl. Cancer Inst. 2003, 95, 1878–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T. BRAF Inhibitors and Melanoma. Cancer J. 2011, 17, 505–511. [Google Scholar] [CrossRef]
- Raaijmakers, M.I.G.; Widmer, D.S.; Narechania, A.; Eichhoff, O.; Freiberger, S.N.; Wenzina, J.; Cheng, P.F.; Mihic-Probst, D.; Desalle, R.; Dummer, R.; et al. Co-Existence of BRAF and NRAS Driver Mutations in the Same Melanoma Cells Results in Heterogeneity of Targeted Therapy Resistance. Oncotarget 2016, 7, 77163–77174. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Vultur, A.; Herlyn, M. Resistance to BRAF Inhibitors: Unraveling Mechanisms and Future Treatment Options. Cancer Res. 2011, 71, 7137–7140. [Google Scholar] [CrossRef] [Green Version]
- Queirolo, P.; Spagnolo, F. BRAF plus MEK-Targeted Drugs: A New Standard of Treatment for BRAF-Mutant Advanced Melanoma. Cancer Metastasis Rev. 2017, 36, 35–42. [Google Scholar] [CrossRef]
- Joneson, T.; Bar-Sagi, D. Ras Effectors and Their Role in Mitogenesis and Oncogenesis. J. Mol. Med. 1997, 75, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Goydos, J.S.; Mann, B.; Kim, H.J.; Gabriel, E.M.; Alsina, J.; Germino, F.J.; Shih, W.; Gorski, D.H. Detection of B-RAF and N-RAS Mutations in Human Melanoma. J. Am. Coll. Surg. 2005, 200, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Omholt, K.; Karsberg, S.; Platz, A.; Kanter, L.; Ringborg, U.; Hansson, J. Screening of N-Ras Codon 61 Mutations in Paired Primary and Metastatic Cutaneous Melanomas: Mutations Occur Early and Persist throughout Tumor Progression. Clin. Cancer Res. 2002, 8, 3468–3474. [Google Scholar]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 Somatic Mutational Landscape in Sporadic Human Cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome Sequencing Identifies Recurrent Mutations in NF1 and RASopathy Genes in Sun-Exposed Melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Lionarons, D.A.; Hancock, D.C.; Rana, S.; East, P.; Moore, C.; Murillo, M.M.; Carvalho, J.; Spencer-Dene, B.; Herbert, E.; Stamp, G.; et al. RAC1P29S Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance. Cancer Cell 2019, 36, 68–83.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome Sequencing Identifies Recurrent Somatic RAC1 Mutations in Melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Goel, V.; Haluska, F.G. PTEN Signaling Pathways in Melanoma. Oncogene 2003, 22, 3113–3122. [Google Scholar] [CrossRef] [Green Version]
- Guldberg, P.; thor Straten, P.; Birck, A.; Ahrenkiel, V.; Kirkin, A.F.; Zeuthen, J. Disruption of the MMAC1/PTEN Gene by Deletion or Mutation Is a Frequent Event in Malignant Melanoma. Cancer Res. 1997, 57, 3660–3663. [Google Scholar] [PubMed]
- Tsao, H.; Zhang, X.; Fowlkes, K.; Haluska, F.G. Relative Reciprocity of NRAS and PTEN/MMAC1 Alterations in Cutaneous Melanoma Cell Lines. Cancer Res. 2000, 60, 1800–1804. [Google Scholar] [PubMed]
- Stahl, J.M.; Cheung, M.; Sharma, A.; Trivedi, N.R.; Shanmugam, S.; Robertson, G.P. Loss of PTEN Promotes Tumor Development in Malignant Melanoma. Cancer Res. 2003, 63, 2881–2890. [Google Scholar] [PubMed]
- Fecher, L.A.; Cummings, S.D.; Keefe, M.J.; Alani, R.M. Toward a Molecular Classification of Melanoma. J. Clin. Oncol. 2007, 25, 1606–1620. [Google Scholar] [CrossRef] [PubMed]
- Monsel, G.; Ortonne, N.; Bagot, M.; Bensussan, A.; Dumaz, N. C-Kit Mutants Require Hypoxia-Inducible Factor 1alpha to Transform Melanocytes. Oncogene 2010, 29, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, S.; Woods, S.L.; Boyle, G.M.; Aoude, L.G.; MacGregor, S.; Zismann, V.; Gartside, M.; Cust, A.E.; Haq, R.; Harland, M.; et al. A Novel Recurrent Mutation in MITF Predisposes to Familial and Sporadic Melanoma. Nature 2011, 480, 99–103. [Google Scholar] [CrossRef]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative Genomic Analyses Identify MITF as a Lineage Survival Oncogene Amplified in Malignant Melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent Somatic Mutations of GNAQ in Uveal Melanoma and Blue Naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [Green Version]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in Uveal Melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [Green Version]
- Nikolaev, S.I.; Rimoldi, D.; Iseli, C.; Valsesia, A.; Robyr, D.; Gehrig, C.; Harshman, K.; Guipponi, M.; Bukach, O.; Zoete, V.; et al. Exome Sequencing Identifies Recurrent Somatic MAP2K1 and MAP2K2 Mutations in Melanoma. Nat. Genet. 2011, 44, 133–139. [Google Scholar] [CrossRef]
- Davies, M.A.; Stemke-Hale, K.; Tellez, C.; Calderone, T.L.; Deng, W.; Prieto, V.G.; Lazar, A.J.F.; Gershenwald, J.E.; Mills, G.B. A Novel AKT3 Mutation in Melanoma Tumours and Cell Lines. Br. J. Cancer 2008, 99, 1265–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, M.S.; Woods, S.L.; Gartside, M.G.; Bonazzi, V.F.; Dutton-Regester, K.; Aoude, L.G.; Chow, D.; Sereduk, C.; Niemi, N.M.; Tang, N.; et al. Frequent Somatic Mutations in MAP3K5 and MAP3K9 in Metastatic Melanoma Identified by Exome Sequencing. Nat. Genet. 2011, 44, 165–169. [Google Scholar] [CrossRef]
- Wei, X.; Walia, V.; Lin, J.C.; Teer, J.K.; Prickett, T.D.; Gartner, J.; Davis, S.; NISC Comparative Sequencing Program; Stemke-Hale, K.; Davies, M.A.; et al. Exome Sequencing Identifies GRIN2A as Frequently Mutated in Melanoma. Nat. Genet. 2011, 43, 442–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma Genome Sequencing Reveals Frequent PREX2 Mutations. Nature 2012, 485, 502–506. [Google Scholar] [CrossRef]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From Mutations to Medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef] [Green Version]
- Koh, J.; Enders, G.H.; Dynlacht, B.D.; Harlow, E. Tumour-Derived P16 Alleles Encoding Proteins Defective in Cell-Cycle Inhibition. Nature 1995, 375, 506–510. [Google Scholar] [CrossRef]
- Hunter, T.; Pines, J. Cyclins and Cancer. II: Cyclin D and CDK Inhibitors Come of Age. Cell 1994, 79, 573–582. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallarelli, A.F.; Rachakonda, P.S.; André, J.; Heidenreich, B.; Riffaud, L.; Bensussan, A.; Kumar, R.; Dumaz, N. TERT Promoter Mutations in Melanoma Render TERT Expression Dependent on MAPK Pathway Activation. Oncotarget 2016, 7, 53127–53136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; de Lichy, M.; Bille, K.; Dessen, P.; d’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-Defective MITF Germline Mutation Predisposes to Melanoma and Renal Carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef]
- Hill, V.K.; Gartner, J.J.; Samuels, Y.; Goldstein, A.M. The Genetics of Melanoma: Recent Advances. Annu. Rev. Genom. Hum. Genet. 2013, 14, 257–279. [Google Scholar] [CrossRef] [Green Version]
- Sturm, R.A.; Box, N.F.; Ramsay, M. Human Pigmentation Genetics: The Difference Is Only Skin Deep. Bioessays 1998, 20, 712–721. [Google Scholar] [CrossRef]
- Chatzinasiou, F.; Lill, C.M.; Kypreou, K.; Stefanaki, I.; Nicolaou, V.; Spyrou, G.; Evangelou, E.; Roehr, J.T.; Kodela, E.; Katsambas, A.; et al. Comprehensive Field Synopsis and Systematic Meta-Analyses of Genetic Association Studies in Cutaneous Melanoma. J. Natl. Cancer Inst. 2011, 103, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Sera, F.; Gandini, S.; Iodice, S.; Caini, S.; Maisonneuve, P.; Fargnoli, M.C. MC1R Variants, Melanoma and Red Hair Color Phenotype: A Meta-Analysis. Int. J. Cancer 2008, 122, 2753–2760. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Joseph, N.M.; Yu, R.; Benhamida, J.; Liu, S.; Prow, T.; Ruben, B.; North, J.; Pincus, L.; Yeh, I.; et al. Genomic and Transcriptomic Analysis Reveals Incremental Disruption of Key Signaling Pathways during Melanoma Evolution. Cancer Cell 2018, 34, 45–55.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic Correlates of Response to CTLA-4 Blockade in Metastatic Melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [Green Version]
- van den Bulk, J.; Verdegaal, E.M.; de Miranda, N.F. Cancer Immunotherapy: Broadening the Scope of Targetable Tumours. Open Biol. 2018, 8, 180037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP Kinase Signalling Pathways in Cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Burotto, M.; Chiou, V.L.; Lee, J.-M.; Kohn, E.C. The MAPK Pathway across Different Malignancies: A New Perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [Green Version]
- Platz, A.; Egyhazi, S.; Ringborg, U.; Hansson, J. Human Cutaneous Melanoma; a Review of NRAS and BRAF Mutation Frequencies in Relation to Histogenetic Subclass and Body Site. Mol. Oncol. 2008, 1, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Mehnert, J.M.; Kluger, H.M. Driver Mutations in Melanoma: Lessons Learned from Bench-to-Bedside Studies. Curr. Oncol. Rep. 2012, 14, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.A. The Role of the PI3K-AKT Pathway in Melanoma. Cancer J. 2012, 18, 142–147. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt Signal Transduction Pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Delmas, V.; Beermann, F.; Martinozzi, S.; Carreira, S.; Ackermann, J.; Kumasaka, M.; Denat, L.; Goodall, J.; Luciani, F.; Viros, A.; et al. Beta-Catenin Induces Immortalization of Melanocytes by Suppressing P16INK4a Expression and Cooperates with N-Ras in Melanoma Development. Genes Dev. 2007, 21, 2923–2935. [Google Scholar] [CrossRef] [Green Version]
- Weeraratna, A.T.; Jiang, Y.; Hostetter, G.; Rosenblatt, K.; Duray, P.; Bittner, M.; Trent, J.M. Wnt5a Signaling Directly Affects Cell Motility and Invasion of Metastatic Melanoma. Cancer Cell 2002, 1, 279–288. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, A.S.; de Almeida, V.H.; Gomes, F.G.; Rezaie, A.R.; Monteiro, R.Q. TR47, a PAR1-Based Peptide, Inhibits Melanoma Cell Migration in Vitro and Metastasis in Vivo. Biochem. Biophys. Res. Commun. 2018, 495, 1300–1304. [Google Scholar] [CrossRef]
- Xu, S.; Tang, J.; Wang, C.; Liu, J.; Fu, Y.; Luo, Y. CXCR7 Promotes Melanoma Tumorigenesis via Src Kinase Signaling. Cell Death Dis. 2019, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- Pla, P.; Larue, L. Involvement of Endothelin Receptors in Normal and Pathological Development of Neural Crest Cells. Int. J. Dev. Biol. 2003, 47, 315–325. [Google Scholar]
- Tafreshi, N.K.; Tichacek, C.J.; Pandya, D.N.; Doligalski, M.L.; Budzevich, M.M.; Kil, H.; Bhatt, N.B.; Kock, N.D.; Messina, J.L.; Ruiz, E.E.; et al. Melanocortin 1 Receptor-Targeted α-Particle Therapy for Metastatic Uveal Melanoma. J. Nucl. Med. 2019, 60, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.L.; Montes de Oca, M.K.; Pal, H.C.; Afaq, F. Potential Therapeutic Targets of Epithelial-Mesenchymal Transition in Melanoma. Cancer Lett. 2017, 391, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Javelaud, D.; Alexaki, V.-I.; Mauviel, A. Transforming Growth Factor-Beta in Cutaneous Melanoma. Pigment Cell Melanoma Res. 2008, 21, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The Clonal Evolution of Tumor Cell Populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem Cells, Cancer, and Cancer Stem Cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A Cell Initiating Human Acute Myeloid Leukaemia after Transplantation into SCID Mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A Human Colon Cancer Cell Capable of Initiating Tumour Growth in Immunodeficient Mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Read, T.-A.; Fogarty, M.P.; Markant, S.L.; McLendon, R.E.; Wei, Z.; Ellison, D.W.; Febbo, P.G.; Wechsler-Reya, R.J. Identification of CD15 as a Marker for Tumor-Propagating Cells in a Mouse Model of Medulloblastoma. Cancer Cell 2009, 15, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinsmith, L.J.; Pierce, G.B. Multipotentiality of Single Embryonal Carcinoma Cells. Cancer Res. 1964, 24, 1544–1551. [Google Scholar]
- Dontu, G.; Al-Hajj, M.; Abdallah, W.M.; Clarke, M.F.; Wicha, M.S. Stem Cells in Normal Breast Development and Breast Cancer. Cell Prolif. 2003, 36 (Suppl. 1), 59–72. [Google Scholar] [CrossRef] [Green Version]
- Eisterer, W.; Jiang, X.; Christ, O.; Glimm, H.; Lee, K.H.; Pang, E.; Lambie, K.; Shaw, G.; Holyoake, T.L.; Petzer, A.L.; et al. Different Subsets of Primary Chronic Myeloid Leukemia Stem Cells Engraft Immunodeficient Mice and Produce a Model of the Human Disease. Leukemia 2005, 19, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, J.E. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates from a Primitive Hematopoietic Cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Parmiani, G. Melanoma Cancer Stem Cells: Markers and Functions. Cancers 2016, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Quintana, E.; Shackleton, M.; Foster, H.R.; Fullen, D.R.; Sabel, M.S.; Johnson, T.M.; Morrison, S.J. Phenotypic Heterogeneity among Tumorigenic Melanoma Cells from Patients That Is Reversible and Not Hierarchically Organized. Cancer Cell 2010, 18, 510–523. [Google Scholar] [CrossRef] [Green Version]
- Ogden, A.T.; Waziri, A.E.; Lochhead, R.A.; Fusco, D.; Lopez, K.; Ellis, J.A.; Kang, J.; Assanah, M.; McKhann, G.M.; Sisti, M.B.; et al. Identification of A2B5+CD133- Tumor-Initiating Cells in Adult Human Gliomas. Neurosurgery 2008, 62, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, K.M.; Kim, S.Y.; Jin, X.; Song, S.Y.; Kong, D.-S.; Lee, J.-I.; Jeon, J.W.; Kim, M.H.; Kang, B.G.; Jung, Y.; et al. Clinical and Biological Implications of CD133-Positive and CD133-Negative Cells in Glioblastomas. Lab. Investig. 2008, 88, 808–815. [Google Scholar] [CrossRef] [Green Version]
- Kath, R.; Jambrosic, J.A.; Holland, L.; Rodeck, U.; Herlyn, M. Development of Invasive and Growth Factor-Independent Cell Variants from Primary Human Melanomas. Cancer Res. 1991, 51, 2205–2211. [Google Scholar] [PubMed]
- Marie, K.L.; Sassano, A.; Yang, H.H.; Michalowski, A.M.; Michael, H.T.; Guo, T.; Tsai, Y.C.; Weissman, A.M.; Lee, M.P.; Jenkins, L.M.; et al. Melanoblast Transcriptome Analysis Reveals Pathways Promoting Melanoma Metastasis. Nat. Commun. 2020, 11, 333. [Google Scholar] [CrossRef] [Green Version]
- Reed, J.A.; Finnerty, B.; Albino, A.P. Divergent Cellular Differentiation Pathways during the Invasive Stage of Cutaneous Malignant Melanoma Progression. Am. J. Pathol. 1999, 155, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Bröcker, E.B.; Magiera, H.; Herlyn, M. Nerve Growth and Expression of Receptors for Nerve Growth Factor in Tumors of Melanocyte Origin. J. Investig. Dermatol. 1991, 96, 662–665. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Hallman, J.; Sangha, N.; Kute, T.E.; Hammarback, J.A.; White, W.L.; Setaluri, V. Expression of Microtubule-Associated Protein 2 in Benign and Malignant Melanocytes: Implications for Differentiation and Progression of Cutaneous Melanoma. Am. J. Pathol. 2001, 158, 2107–2115. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Pinner, S.; Jordan, P.; Sharrock, K.; Bazley, L.; Collinson, L.; Marais, R.; Bonvin, E.; Goding, C.; Sahai, E. Intravital Imaging Reveals Transient Changes in Pigment Production and Brn2 Expression during Metastatic Melanoma Dissemination. Cancer Res. 2009, 69, 7969–7977. [Google Scholar] [CrossRef] [Green Version]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A Temporarily Distinct Subpopulation of Slow-Cycling Melanoma Cells Is Required for Continuous Tumor Growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.K.; Manglik, V.; Elias, E.G. Immuno-Expression of Human Melanoma Stem Cell Markers in Tissues at Different Stages of the Disease. J. Surg. Res. 2010, 163, e11–e15. [Google Scholar] [CrossRef]
- Sarrió, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-Mesenchymal Transition in Breast Cancer Relates to the Basal-like Phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingber, D.E. Can Cancer Be Reversed by Engineering the Tumor Microenvironment? Semin. Cancer Biol. 2008, 18, 356–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marjanovic, N.D.; Weinberg, R.A.; Chaffer, C.L. Cell Plasticity and Heterogeneity in Cancer. Clin. Chem. 2013, 59, 168–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient Tumour Formation by Single Human Melanoma Cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Döbbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In Vivo Switching of Human Melanoma Cells between Proliferative and Invasive States. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef] [Green Version]
- Hoek, K.S.; Schlegel, N.C.; Brafford, P.; Sucker, A.; Ugurel, S.; Kumar, R.; Weber, B.L.; Nathanson, K.L.; Phillips, D.J.; Herlyn, M.; et al. Metastatic Potential of Melanomas Defined by Specific Gene Expression Profiles with No BRAF Signature. Pigment Cell Res. 2006, 19, 290–302. [Google Scholar] [CrossRef]
- Hoek, K.S.; Goding, C.R. Cancer Stem Cells versus Phenotype-Switching in Melanoma. Pigment Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef]
- Eichhoff, O.M.; Zipser, M.C.; Xu, M.; Weeraratna, A.T.; Mihic, D.; Dummer, R.; Hoek, K.S. The Immunohistochemistry of Invasive and Proliferative Phenotype Switching in Melanoma: A Case Report. Melanoma Res. 2010, 20, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, S.; Kashima, Y.; Suzuki, A.; Suzuki, Y. Single-Cell and Spatial Analyses of Cancer Cells: Toward Elucidating the Molecular Mechanisms of Clonal Evolution and Drug Resistance Acquisition. Inflamm. Regen. 2021, 41, 22. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffs, A.R.; Glover, A.C.; Slobbe, L.J.; Wang, L.; He, S.; Hazlett, J.A.; Awasthi, A.; Woolley, A.G.; Marshall, E.S.; Joseph, W.R.; et al. A Gene Expression Signature of Invasive Potential in Metastatic Melanoma Cells. PLoS ONE 2009, 4, e8461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thrane, K.; Eriksson, H.; Maaskola, J.; Hansson, J.; Lundeberg, J. Spatially Resolved Transcriptomics Enables Dissection of Genetic Heterogeneity in Stage III Cutaneous Malignant Melanoma. Cancer Res. 2018, 78, 5970–5979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, E.S.; Matthews, J.H.; Shaw, J.H.; Nixon, J.; Tumewu, P.; Finlay, G.J.; Holdaway, K.M.; Baguley, B.C. Radiosensitivity of New and Established Human Melanoma Cell Lines: Comparison of [3H]Thymidine Incorporation and Soft Agar Clonogenic Assays. Eur. J. Cancer 1994, 30, 1370–1376. [Google Scholar] [CrossRef]
- D’mello, S.A.N.; Flanagan, J.U.; Green, T.N.; Leung, E.Y.; Askarian-Amiri, M.E.; Joseph, W.R.; McCrystal, M.R.; Isaacs, R.J.; Shaw, J.H.F.; Furneaux, C.E.; et al. Evidence That GRIN2A Mutations in Melanoma Correlate with Decreased Survival. Front. Oncol. 2014, 3, 333. [Google Scholar] [CrossRef] [Green Version]
- D’mello, S.A.N.; Joseph, W.R.; Green, T.N.; Leung, E.Y.; During, M.J.; Finlay, G.J.; Baguley, B.C.; Kalev-Zylinska, M.L. Selected GRIN2A Mutations in Melanoma Cause Oncogenic Effects That Can Be Modulated by Extracellular Glutamate. Cell Calcium 2016, 60, 384–395. [Google Scholar] [CrossRef]
- Motwani, J.; Rodger, E.J.; Stockwell, P.A.; Baguley, B.C.; Macaulay, E.C.; Eccles, M.R. Genome-Wide DNA Methylation and RNA Expression Differences Correlate with Invasiveness in Melanoma Cell Lines. Epigenomics 2021, 13, 577–598. [Google Scholar] [CrossRef]
- Tran, K.B.; Kolekar, S.; Jabed, A.; Jaynes, P.; Shih, J.-H.; Wang, Q.; Flanagan, J.U.; Rewcastle, G.W.; Baguley, B.C.; Shepherd, P.R. Diverse Mechanisms Activate the PI 3-Kinase/MTOR Pathway in Melanomas: Implications for the Use of PI 3-Kinase Inhibitors to Overcome Resistance to Inhibitors of BRAF and MEK. BMC Cancer 2021, 21, 136. [Google Scholar] [CrossRef]
- Tran, K.B.; Gimenez, G.; Tsai, P.; Kolekar, S.; Rodger, E.J.; Chatterjee, A.; Jabed, A.; Shih, J.-H.; Joseph, W.R.; Marshall, E.S.; et al. Genomic and Signalling Pathway Characterization of the NZM Panel of Melanoma Cell Lines: A Valuable Model for Studying the Impact of Genetic Diversity in Melanoma. Pigment Cell Melanoma Res. 2021, 34, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Finlay, G.J.; Baguley, B.C. The Role of the Hippo Pathway in Melanocytes and Melanoma. Front. Oncol. 2013, 3, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Leung, E.; Baguley, B.C.; Finlay, G.J. Heterogeneity of Expression of Epithelial-Mesenchymal Transition Markers in Melanocytes and Melanoma Cell Lines. Front. Genet. 2013, 4, 97. [Google Scholar] [CrossRef] [Green Version]
- Rodger, E.J.; Almomani, S.N.; Ludgate, J.L.; Stockwell, P.A.; Baguley, B.C.; Eccles, M.R.; Chatterjee, A. Comparison of Global DNA Methylation Patterns in Human Melanoma Tissues and Their Derivative Cell Lines. Cancers 2021, 13, 2123. [Google Scholar] [CrossRef]
- Eccles, M.R.; He, S.; Ahn, A.; Slobbe, L.J.; Jeffs, A.R.; Yoon, H.-S.; Baguley, B.C. MITF and PAX3 Play Distinct Roles in Melanoma Cell Migration; Outline of a “Genetic Switch” Theory Involving MITF and PAX3 in Proliferative and Invasive Phenotypes of Melanoma. Front. Oncol. 2013, 3, 229. [Google Scholar] [CrossRef] [Green Version]
- Charters, G.A.; Stones, C.J.; Shelling, A.N.; Baguley, B.C.; Finlay, G.J. Centrosomal Dysregulation in Human Metastatic Melanoma Cell Lines. Cancer Genet. 2011, 204, 477–485. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Li, C.G.; Slobbe, L.; Glover, A.; Marshall, E.; Baguley, B.C.; Eccles, M.R. PAX3 Knockdown in Metastatic Melanoma Cell Lines Does Not Reduce MITF Expression. Melanoma Res. 2011, 21, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Stones, C.J.; Kim, J.E.; Joseph, W.R.; Leung, E.; Marshall, E.S.; Finlay, G.J.; Shelling, A.N.; Baguley, B.C. Comparison of Responses of Human Melanoma Cell Lines to MEK and BRAF Inhibitors. Front. Genet. 2013, 4, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granados, K.; Poelchen, J.; Novak, D.; Utikal, J. Cellular Reprogramming-A Model for Melanoma Cellular Plasticity. Int. J. Mol. Sci. 2020, 21, 8274. [Google Scholar] [CrossRef]
- Chatterjee, A.; Stockwell, P.A.; Ahn, A.; Rodger, E.J.; Leichter, A.L.; Eccles, M.R. Genome-Wide Methylation Sequencing of Paired Primary and Metastatic Cell Lines Identifies Common DNA Methylation Changes and a Role for EBF3 as a Candidate Epigenetic Driver of Melanoma Metastasis. Oncotarget 2017, 8, 6085–6101. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-Stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5. [Google Scholar] [CrossRef] [Green Version]
- Ahn, A.; Rodger, E.J.; Motwani, J.; Gimenez, G.; Stockwell, P.A.; Parry, M.; Hersey, P.; Chatterjee, A.; Eccles, M.R. Transcriptional Reprogramming and Constitutive PD-L1 Expression in Melanoma Are Associated with Dedifferentiation and Activation of Interferon and Tumour Necrosis Factor Signalling Pathways. Cancers 2021, 13, 4250. [Google Scholar] [CrossRef]
- Widmer, D.S.; Hoek, K.S.; Cheng, P.F.; Eichhoff, O.M.; Biedermann, T.; Raaijmakers, M.I.G.; Hemmi, S.; Dummer, R.; Levesque, M.P. Hypoxia Contributes to Melanoma Heterogeneity by Triggering HIF1α-Dependent Phenotype Switching. J. Investig. Dermatol. 2013, 133, 2436–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucciarelli, D.; Lengger, N.; Takáčová, M.; Csaderova, L.; Bartosova, M.; Breiteneder, H.; Pastorekova, S.; Hafner, C. Hypoxia Increases the Heterogeneity of Melanoma Cell Populations and Affects the Response to Vemurafenib. Mol. Med. Rep. 2016, 13, 3281–3288. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wölfel, T.; Hölzel, M.; et al. Melanomas Resist T-Cell Therapy through Inflammation-Induced Reversible Dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Hölzel, M.; Tüting, T. Inflammation-Induced Plasticity in Melanoma Therapy and Metastasis. Trends Immunol. 2016, 37, 364–374. [Google Scholar] [CrossRef]
- Feige, E.; Yokoyama, S.; Levy, C.; Khaled, M.; Igras, V.; Lin, R.J.; Lee, S.; Widlund, H.R.; Granter, S.R.; Kung, A.L.; et al. Hypoxia-Induced Transcriptional Repression of the Melanoma-Associated Oncogene MITF. Proc. Natl. Acad. Sci. USA 2011, 108, E924–E933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Santinon, F.; Flores González, R.E.; del Rincón, S.V. Melanoma Plasticity: Promoter of Metastasis and Resistance to Therapy. Front. Oncol. 2021, 11, 756001. [Google Scholar] [CrossRef]
- Knappe, N.; Novak, D.; Weina, K.; Bernhardt, M.; Reith, M.; Larribere, L.; Hölzel, M.; Tüting, T.; Gebhardt, C.; Umansky, V.; et al. Directed Dedifferentiation Using Partial Reprogramming Induces Invasive Phenotype in Melanoma Cells. Stem Cells 2016, 34, 832–846. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Zhao, B.; Li, D.; Yin, G. Long Non-Coding RNA CASC15 Promotes Melanoma Progression by Epigenetically Regulating PDCD4. Cell Biosci. 2018, 8, 42. [Google Scholar] [CrossRef]
- Diener, J.; Baggiolini, A.; Pernebrink, M.; Dalcher, D.; Lerra, L.; Cheng, P.F.; Varum, S.; Häusel, J.; Stierli, S.; Treier, M.; et al. Epigenetic Control of Melanoma Cell Invasiveness by the Stem Cell Factor SALL4. Nat. Commun. 2021, 12, 5056. [Google Scholar] [CrossRef]
- Rambow, F.; Bechadergue, A.; Luciani, F.; Gros, G.; Domingues, M.; Bonaventure, J.; Meurice, G.; Marine, J.-C.; Larue, L. Regulation of Melanoma Progression through the TCF4/MiR-125b/NEDD9 Cascade. J. Investig. Dermatol. 2016, 136, 1229–1237. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, J.N.; Aguero, T.H.; Owens, D.A.; Kurtenbach, S.; Field, M.G.; Durante, M.A.; Rodriguez, D.A.; King, M.L.; Harbour, J.W. BAP1 Regulates Epigenetic Switch from Pluripotency to Differentiation in Developmental Lineages Giving Rise to BAP1-Mutant Cancers. Sci. Adv. 2019, 5, eaax1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.F.; Shakhova, O.; Widmer, D.S.; Eichhoff, O.M.; Zingg, D.; Frommel, S.C.; Belloni, B.; Raaijmakers, M.I.; Goldinger, S.M.; Santoro, R.; et al. Methylation-Dependent SOX9 Expression Mediates Invasion in Human Melanoma Cells and Is a Negative Prognostic Factor in Advanced Melanoma. Genome Biol. 2015, 16, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fane, M.E.; Chhabra, Y.; Hollingsworth, D.E.J.; Simmons, J.L.; Spoerri, L.; Oh, T.G.; Chauhan, J.; Chin, T.; Harris, L.; Harvey, T.J.; et al. NFIB Mediates BRN2 Driven Melanoma Cell Migration and Invasion Through Regulation of EZH2 and MITF. EBioMedicine 2017, 16, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Ghislin, S.; Deshayes, F.; Middendorp, S.; Boggetto, N.; Alcaide-Loridan, C. PHF19 and Akt Control the Switch between Proliferative and Invasive States in Melanoma. Cell Cycle 2012, 11, 1634–1645. [Google Scholar] [CrossRef] [Green Version]
- Emmons, M.F.; Faião-Flores, F.; Sharma, R.; Thapa, R.; Messina, J.L.; Becker, J.C.; Schadendorf, D.; Seto, E.; Sondak, V.K.; Koomen, J.M.; et al. HDAC8 Regulates a Stress Response Pathway in Melanoma to Mediate Escape from BRAF Inhibitor Therapy. Cancer Res. 2019, 79, 2947–2961. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Smalley, I.; Emmons, M.F.; Sharma, R.; Izumi, V.; Messina, J.; Koomen, J.M.; Pasquale, E.B.; Forsyth, P.A.; Smalley, K.S.M. Noncanonical EphA2 Signaling Is a Driver of Tumor-Endothelial Cell Interactions and Metastatic Dissemination in BRAF Inhibitor‒Resistant Melanoma. J. Investig. Dermatol. 2021, 141, 840–851.e4. [Google Scholar] [CrossRef]
- Moran, B.; Silva, R.; Perry, A.S.; Gallagher, W.M. Epigenetics of Malignant Melanoma. Semin. Cancer Biol. 2018, 51, 80–88. [Google Scholar] [CrossRef]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the Regulatory Landscape of Melanoma Reveals TEADS as Regulators of the Invasive Cell State. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [Green Version]
- Manning, C.S.; Hooper, S.; Sahai, E.A. Intravital Imaging of SRF and Notch Signalling Identifies a Key Role for EZH2 in Invasive Melanoma Cells. Oncogene 2015, 34, 4320–4332. [Google Scholar] [CrossRef] [Green Version]
- Tiffen, J.; Gallagher, S.J.; Hersey, P. EZH2: An Emerging Role in Melanoma Biology and Strategies for Targeted Therapy. Pigment Cell Melanoma Res. 2015, 28, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Tiffen, J.; Gallagher, S.J.; Filipp, F.; Gunatilake, D.; Emran, A.A.; Cullinane, C.; Dutton-Register, K.; Aoude, L.; Hayward, N.; Chatterjee, A.; et al. EZH2 Cooperates with DNA Methylation to Downregulate Key Tumor Suppressors and IFN Gene Signatures in Melanoma. J. Investig. Dermatol. 2020, 140, 2442–2454.e5. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Biondo, C.; Lentini, M.; Catalano, T.; Teti, D.; Venza, I. Epigenetic Regulation of P14ARF and P16INK4A Expression in Cutaneous and Uveal Melanoma. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 247–256. [Google Scholar] [CrossRef]
- Jonsson, A.; Tuominen, R.; Grafström, E.; Hansson, J.; Egyhazi, S. High Frequency of P16INK4A Promoter Methylation in NRAS-Mutated Cutaneous Melanoma. J. Investig. Dermatol. 2010, 130, 2809–2817. [Google Scholar] [CrossRef] [Green Version]
- Hoon, D.S.B.; Spugnardi, M.; Kuo, C.; Huang, S.K.; Morton, D.L.; Taback, B. Profiling Epigenetic Inactivation of Tumor Suppressor Genes in Tumors and Plasma from Cutaneous Melanoma Patients. Oncogene 2004, 23, 4014–4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirmohammadsadegh, A.; Marini, A.; Nambiar, S.; Hassan, M.; Tannapfel, A.; Ruzicka, T.; Hengge, U.R. Epigenetic Silencing of the PTEN Gene in Melanoma. Cancer Res. 2006, 66, 6546–6552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahtz, C.; Stranzenbach, R.; Fiedler, E.; Helmbold, P.; Dammann, R.H. Methylation of PTEN as a Prognostic Factor in Malignant Melanoma of the Skin. J. Investig. Dermatol. 2010, 130, 620–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.-H.; Lok, H.C.; Sahni, S.; Lane, D.J.R.; Richardson, D.R. Roads to Melanoma: Key Pathways and Emerging Players in Melanoma Progression and Oncogenic Signaling. Biochim. Biophys. Acta 2016, 1863, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.-Y.; Kandel, J.J.; Yamashiro, D.J.; Canoll, P.; Anastassiou, D. A Multi-Cancer Mesenchymal Transition Gene Expression Signature Is Associated with Prolonged Time to Recurrence in Glioblastoma. PLoS ONE 2012, 7, e34705. [Google Scholar] [CrossRef]
- Kim, H.; Watkinson, J.; Varadan, V.; Anastassiou, D. Multi-Cancer Computational Analysis Reveals Invasion-Associated Variant of Desmoplastic Reaction Involving INHBA, THBS2 and COL11A1. BMC Med. Genom. 2010, 3, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Péterfi, Z.; Donkó, A.; Orient, A.; Sum, A.; Prókai, A.; Molnár, B.; Veréb, Z.; Rajnavölgyi, E.; Kovács, K.J.; Müller, V.; et al. Peroxidasin Is Secreted and Incorporated into the Extracellular Matrix of Myofibroblasts and Fibrotic Kidney. Am. J. Pathol. 2009, 175, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Jayachandran, A.; Prithviraj, P.; Lo, P.-H.; Walkiewicz, M.; Anaka, M.; Woods, B.L.; Tan, B.; Behren, A.; Cebon, J.; McKeown, S.J. Identifying and Targeting Determinants of Melanoma Cellular Invasion. Oncotarget 2016, 7, 41186–41202. [Google Scholar] [CrossRef] [Green Version]
- Ye, I.C.; Fertig, E.J.; DiGiacomo, J.W.; Considine, M.; Godet, I.; Gilkes, D.M. Molecular Portrait of Hypoxia in Breast Cancer: A Prognostic Signature and Novel HIF-Regulated Genes. Mol. Cancer Res. 2018, 16, 1889–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Guo, X.; Zhang, X.; Zhou, W.; Liu, Z.; Wang, J.; Zhang, T.; Mao, Z.; Luo, J.; Jin, T.; et al. SYNJ2 Variant Rs9365723 Is Associated with Colorectal Cancer Risk in Chinese Han Population. Int. J. Biol. Markers 2016, 31, e138–e143. [Google Scholar] [CrossRef]
- Ben-Chetrit, N.; Chetrit, D.; Russell, R.; Körner, C.; Mancini, M.; Abdul-Hai, A.; Itkin, T.; Carvalho, S.; Cohen-Dvashi, H.; Koestler, W.J.; et al. Synaptojanin 2 Is a Druggable Mediator of Metastasis and the Gene Is Overexpressed and Amplified in Breast Cancer. Sci. Signal 2015, 8, ra7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Li, L.; Jiang, G.; Zhan, H.; Wang, N. B-Cell CLL/Lymphoma 3 Promotes Glioma Cell Proliferation and Inhibits Apoptosis through the Oncogenic STAT3 Pathway. Int. J. Oncol. 2016, 49, 2471–2479. [Google Scholar] [CrossRef] [Green Version]
- Legge, D.N.; Shephard, A.P.; Collard, T.J.; Greenhough, A.; Chambers, A.C.; Clarkson, R.W.; Paraskeva, C.; Williams, A.C. BCL-3 Promotes a Cancer Stem Cell Phenotype by Enhancing β-Catenin Signalling in Colorectal Tumour Cells. Dis. Models Mech. 2019, 12, dmm037697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.M.; Shin, H.; Choi, W.; Ahn, S.; Kim, W. Characterization of Human SMARCE1r High-Mobility-Group Protein. Biochim. Biophys. Acta 2002, 1574, 269–276. [Google Scholar] [CrossRef]
- Lee, M.; Daniels, M.J.; Garnett, M.J.; Venkitaraman, A.R. A Mitotic Function for the High-Mobility Group Protein HMG20b Regulated by Its Interaction with the BRC Repeats of the BRCA2 Tumor Suppressor. Oncogene 2011, 30, 3360–3369. [Google Scholar] [CrossRef] [Green Version]
- Rountree, M.R.; Bachman, K.E.; Baylin, S.B. DNMT1 Binds HDAC2 and a New Co-Repressor, DMAP1, to Form a Complex at Replication Foci. Nat. Genet. 2000, 25, 269–277. [Google Scholar] [CrossRef]
- Ismail, T.; Lee, H.-K.; Kim, C.; Kwon, T.; Park, T.J.; Lee, H.-S. KDM1A Microenvironment, Its Oncogenic Potential, and Therapeutic Significance. Epigenet. Chromatin 2018, 11, 33. [Google Scholar] [CrossRef]
- Blank, J.L.; Liu, X.J.; Cosmopoulos, K.; Bouck, D.C.; Garcia, K.; Bernard, H.; Tayber, O.; Hather, G.; Liu, R.; Narayanan, U.; et al. Novel DNA Damage Checkpoints Mediating Cell Death Induced by the NEDD8-Activating Enzyme Inhibitor MLN4924. Cancer Res. 2013, 73, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiessling, A.A.; Bletsa, R.; Desmarais, B.; Mara, C.; Kallianidis, K.; Loutradis, D. Genome-Wide Microarray Evidence That 8-Cell Human Blastomeres over-Express Cell Cycle Drivers and under-Express Checkpoints. J. Assist. Reprod. Genet. 2010, 27, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicod, M.; Michlig, S.; Flahaut, M.; Salinas, M.; Fowler Jaeger, N.; Horisberger, J.-D.; Rossier, B.C.; Firsov, D. A Novel Vasopressin-Induced Transcript Promotes MAP Kinase Activation and ENaC Downregulation. EMBO J. 2002, 21, 5109–5117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch-Sutherland, C.F.; Chatterjee, A.; Stockwell, P.A.; Eccles, M.R.; Macaulay, E.C. Reawakening the Developmental Origins of Cancer Through Transposable Elements. Front. Oncol. 2020, 10, 468. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motwani, J.; Eccles, M.R. Genetic and Genomic Pathways of Melanoma Development, Invasion and Metastasis. Genes 2021, 12, 1543. https://doi.org/10.3390/genes12101543
Motwani J, Eccles MR. Genetic and Genomic Pathways of Melanoma Development, Invasion and Metastasis. Genes. 2021; 12(10):1543. https://doi.org/10.3390/genes12101543
Chicago/Turabian StyleMotwani, Jyoti, and Michael R. Eccles. 2021. "Genetic and Genomic Pathways of Melanoma Development, Invasion and Metastasis" Genes 12, no. 10: 1543. https://doi.org/10.3390/genes12101543
APA StyleMotwani, J., & Eccles, M. R. (2021). Genetic and Genomic Pathways of Melanoma Development, Invasion and Metastasis. Genes, 12(10), 1543. https://doi.org/10.3390/genes12101543