
Chronological Incongruences between Mitochondrial and Nuclear Phylogenies of Aedes Mosquitoes




Abstract
:1. Introduction
2. Materials and Methods
2.1. Genes and Taxa Selection
2.2. Alignments and Phylogenetic Analyses
2.3. Divergence Estimates
3. Results
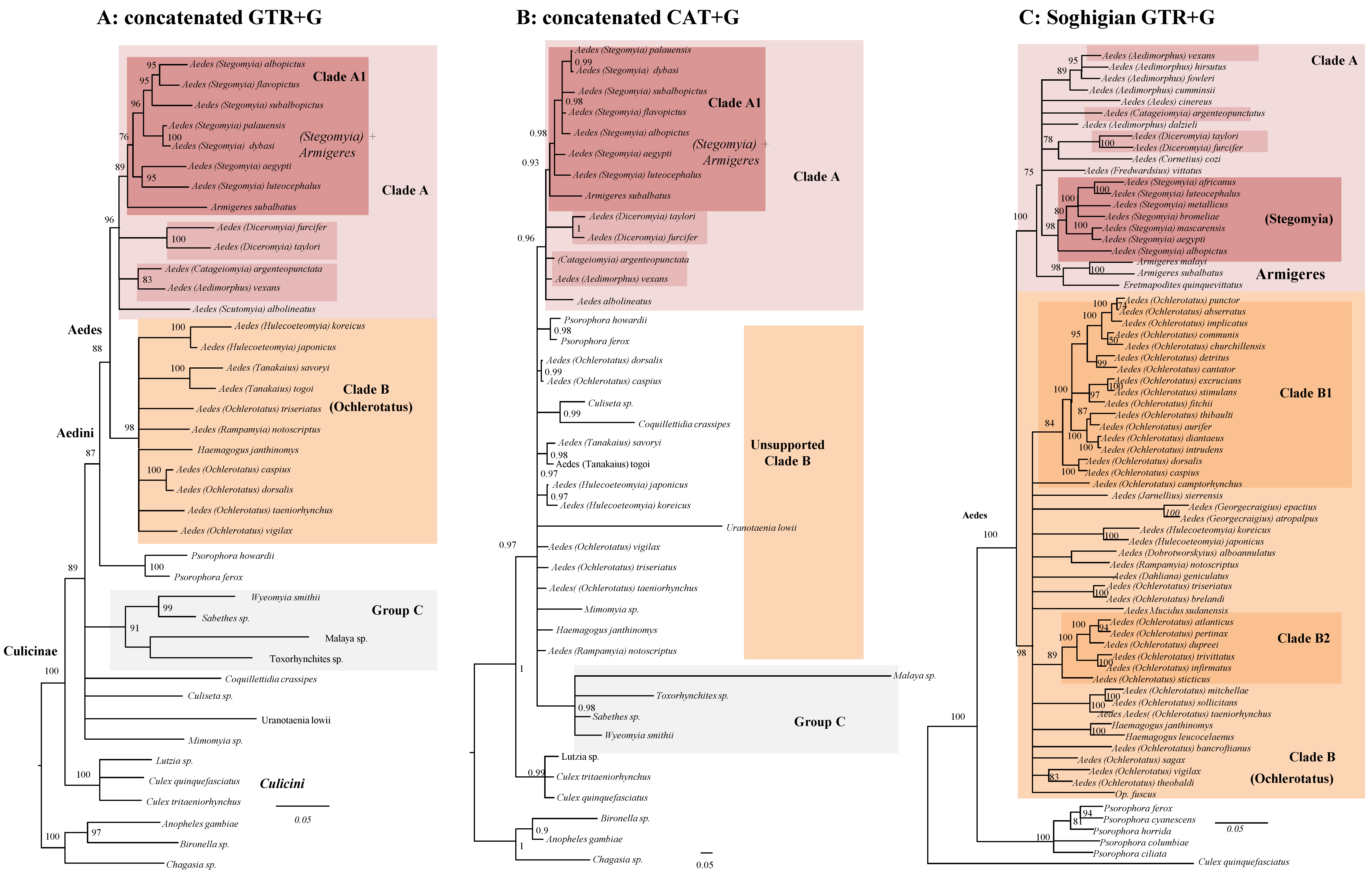
3.1. Conflicts between Nuclear and Mitochondrial Phylogenies
3.2. A Conservative Picture of Aedini and Other Culicinae Phylogeny
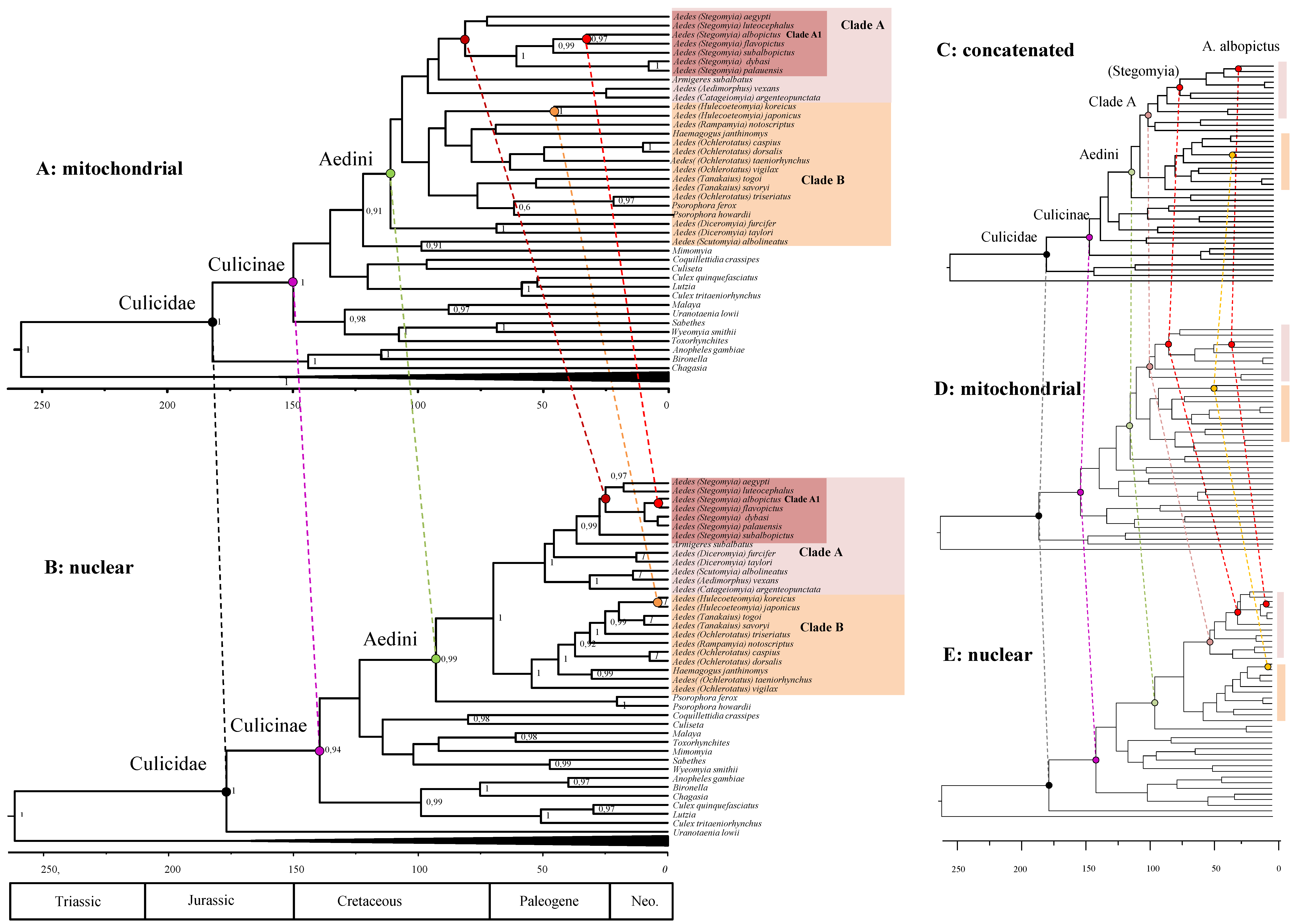
3.3. Divergence Estimates of the Aedini
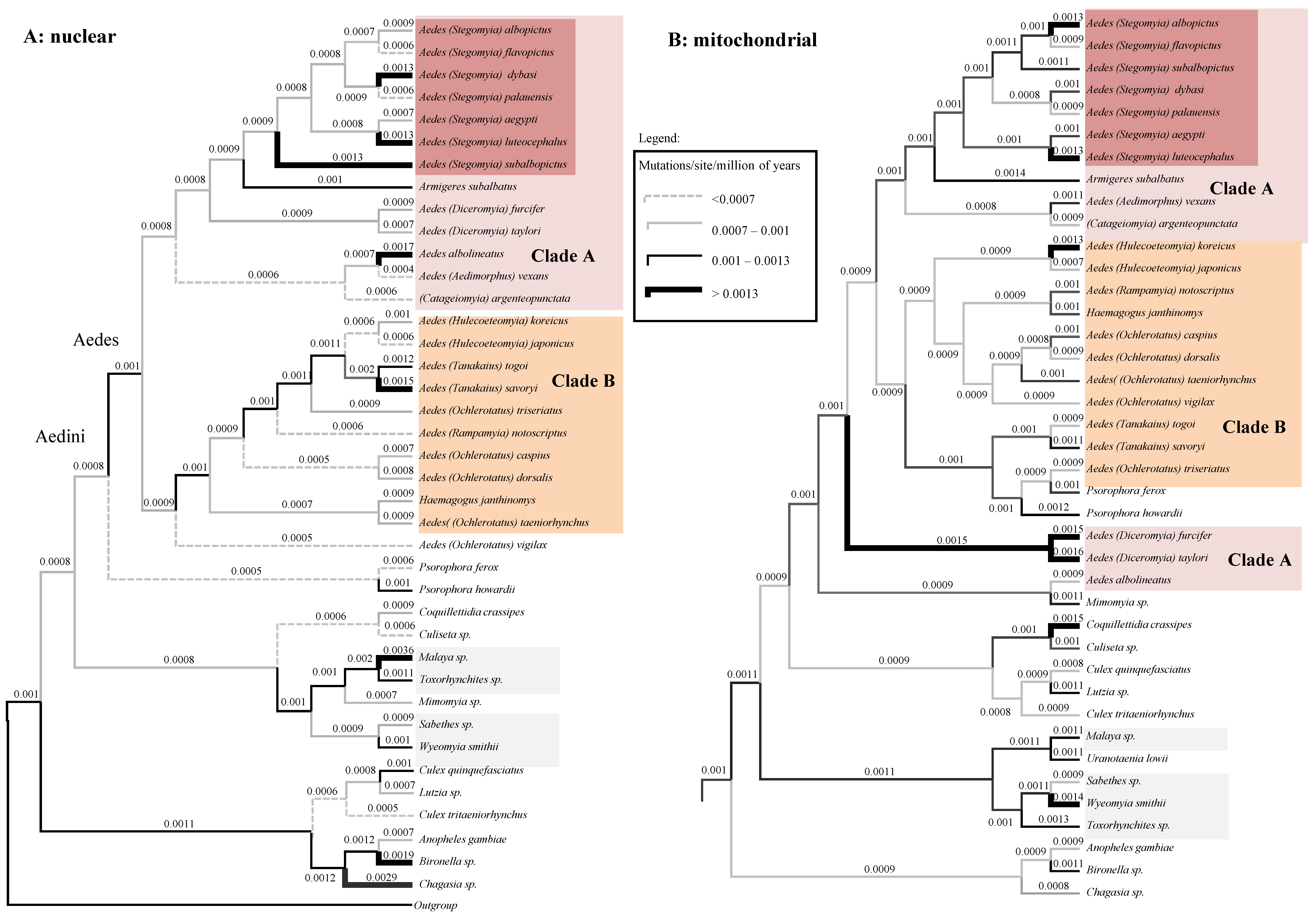
3.4. Chronological Incongruences between Nuclear and Mitochondrial Data
3.5. Mitochondrial-Nuclear Chronological Incongruences Are Consistent over Different Analytical Condition
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reinert, J.F.; Harbach, R.E.; Kitching, I.J. Phylogeny and Classification of Aedini (Diptera: Culicidae), Based on Morphological Characters of All Life Stages. Zool. J. Linn. Soc. 2004, 142, 289–368. [Google Scholar] [CrossRef] [Green Version]
- Pombi, M.; Montarsi, F. Mosquitoes (Culicidae). In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Wilkerson, R.C.; Linton, Y.M.; Fonseca, D.M.; Schultz, T.R.; Price, D.C.; Strickman, D.A. Making Mosquito Taxonomy Useful: A Stable Classification of Tribe Aedini That Balances Utility with Current Knowledge of Evolutionary Relationships. PLoS ONE 2015, 10, e0133602. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.J.; Hoerauf, A.; Bockarie, M. Lymphatic Filariasis and Onchocerciasis. Lancet 2010, 376, 1175–1185. [Google Scholar] [CrossRef]
- Pfeffer, M.; Dobler, G. Emergence of Zoonotic Arboviruses by Animal Trade and Migration. Parasites Vectors 2010, 3, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverj, A.; Rota-Stabelli, O. On the correct interpretation of similarity index in codon usage studies: Comparison with four other metrics and implications for Zika and West Nile virus. Virus Res. 2020, 286, 198097. [Google Scholar] [CrossRef]
- Kraemer, M.U.G.; Sinka, M.E.; Duda, K.A.; Mylne, A.Q.N.; Shearer, F.M.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; Coelho, G.E.; Van Bortel, W.; et al. The Global Distribution of the Arbovirus Vectors Aedes Aegypti and Ae. Albopictus. eLife 2015, 4, e08347. [Google Scholar] [CrossRef]
- Medlock, J.M.; Hansford, K.M.; Schaffner, F.; Versteirt, V.; Hendrickx, G.; Zeller, H.; Van Bortel, W. A Review of the Invasive Mosquitoes in Europe: Ecology, Public Health Risks, and Control Options. Vector-Borne Zoonotic Dis. 2012, 12, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Cameron, E.C.; Wilkerson, R.C.; Mogi, M.; Miyagi, I.; Toma, T.; Kim, H.C.; Fonseca, D.M. Molecular Phylogenetics of Aedes Japonicus, a Disease Vector That Recently Invaded Western Europe, North America, and the Hawaiian Islands. J. Med. Entomol. 2010, 47, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Medlock, J.M.; Hansford, K.M.; Versteirt, V.; Cull, B.; Kampen, H.; Fontenille, D.; Hendrickx, G.; Zeller, H.; Van Bortel, W.; Schaffner, F. An Entomological Review of Invasive Mosquitoes in Europe. Bull. Entomol. Res. 2015, 105, 637–663. [Google Scholar] [CrossRef] [PubMed]
- Grard, G.; Moureau, G.; Charrel, R.N.; Holmes, E.C.; Gould, E.A.; de Lamballerie, X. Genomics and Evolution of Aedes-Borne Flaviviruses. J. Gen. Virol. 2010, 91, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, F.; Chouin, S.; Guilloteau, J. First Record of Ochlerotatus (Finlaya) Japonicus Japonicus (Theobald, 1901) in Metropolitan France. J. Am. Mosq. Control Assoc. 2003, 19, 1–5. [Google Scholar] [PubMed]
- Faria, N.R.; Quick, J.; Claro, I.M.; Thézé, J.; De Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.G.; Hill, S.C.; Black, A.; Da Costa, A.C.; et al. Establishment and Cryptic Transmission of Zika Virus in Brazil and the Americas. Nature 2017, 546, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-G.; Jiang, X.; Gu, J.; Xu, M.; Wu, Y.; Deng, Y.; Zhang, C.; Bonizzoni, M.; Dermauw, W.; Vontas, J.; et al. Genome Sequence of the Asian Tiger Mosquito, Aedes Albopictus, Reveals Insights into Its Biology, Genetics, and Evolution. Proc. Natl. Acad. Sci. USA 2015, 112, e5907–e5915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, B.J.; Dudchenko, O.; Kingan, S.B.; Koren, S.; Antoshechkin, I.; Crawford, J.E.; Glassford, W.J.; Herre, M.; Redmond, S.N.; Rose, N.H.; et al. Improved Reference Genome of Aedes Aegypti Informs Arbovirus Vector Control. Nature 2018, 563, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Dritsou, V.; Topalis, P.; Windbichler, N.; Simoni, A.; Hall, A.; Lawson, D.; Hinsley, M.; Hughes, D.; Napolioni, V.; Crucianelli, F.; et al. A Draft Genome Sequence of an Invasive Mosquito: An Italian Aedes Albopictus. Pathog. Glob. Health 2015, 109, 207–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, B.; Montarsi, F.; Huemer, H.P.; Indra, A.; Capelli, G.; Allerberger, F.; Nowotny, N. First Record of the Asian Bush Mosquito, Aedes Japonicus Japonicus, in Italy: Invasion from an Established Austrian Population. Parasites Vectors 2016, 9, 284. [Google Scholar] [CrossRef] [Green Version]
- Montarsi, F.; Drago, A.; Martini, S.; Calzolari, M.; De Filippo, F.; Bianchi, A.; Mazzucato, M.; Ciocchetta, S.; Arnoldi, D.; Baldacchino, F.; et al. Current Distribution of the Invasive Mosquito Species, Aedes Koreicus [Hulecoeteomyia Koreica] in Northern Italy. Parasites Vectors 2015, 8, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Capelli, G.; Drago, A.; Martini, S.; Montarsi, F.; Soppelsa, M.; Delai, N.; Ravagnan, S.; Mazzon, L.; Schaffner, F.; Mathis, A.; et al. First Report in Italy of the Exotic Mosquito Species Aedes (Finlaya) Koreicus, a Potential Vector of Arboviruses and Filariae. Parasites Vectors 2011, 4, 188. [Google Scholar] [CrossRef] [Green Version]
- Huber, K.; Jansen, S.; Leggewie, M.; Badusche, M.; Schmidt-Chanasit, J.; Becker, N.; Tannich, E.; Becker, S.C. Aedes Japonicus Japonicus (Diptera: Culicidae) from Germany Have Vector Competence for Japan Encephalitis Virus but Are Refractory to Infection with West Nile Virus. Parasitol. Res. 2014, 113, 3195–3199. [Google Scholar] [CrossRef]
- Soghigian, J.; Andreadis, T.G.; Livdahl, T.P. From Ground Pools to Treeholes: Convergent Evolution of Habitat and Phenotype in Aedes Mosquitoes. BMC Evol. Biol. 2017, 17, 262. [Google Scholar] [CrossRef]
- Ramasamy, S.; Ometto, L.; Crava, C.M.; Revadi, S.; Kaur, R.; Horner, D.S.; Pisani, D.; Dekker, T.; Anfora, G.; Rota-Stabelli, O. The Evolution of Olfactory Gene Families in Drosophila and the Genomic Basis of Chemical-Ecological Adaptation in Drosophila Suzukii. Genome Biol. Evol. 2016, 8, 2297–2311. [Google Scholar] [CrossRef] [Green Version]
- Crava, C.M.; Brütting, C.; Baldwin, I.T. Transcriptome Profiling Reveals Differential Gene Expression of Detoxification Enzymes in a Hemimetabolous Tobacco Pest after Feeding on Jasmonate-Silenced Nicotiana Attenuata Plants. BMC Genom. 2016, 17, 1005. [Google Scholar] [CrossRef] [Green Version]
- Ometto, L.; Cestaro, A.; Ramasamy, S.; Grassi, A.; Revadi, S.; Siozios, S.; Moretto, M.; Fontana, P.; Varotto, C.; Pisani, D.; et al. Linking Genomics and Ecology to Investigate the Complex Evolution of an Invasive Drosophila Pest. Genome Biol. Evol. 2013, 5, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Rota-Stabelli, O.; Ometto, L.; Tait, G.; Ghirotto, S.; Kaur, R.; Drago, F.; González, J.; Walton, V.M.; Anfora, G.; Rossi-Stacconi, M.V. Distinct Genotypes and Phenotypes in European and American Strains of Drosophila Suzukii: Implications for Biology and Management of an Invasive Organism. J. Pest Sci. 2020, 93, 77–89. [Google Scholar] [CrossRef]
- Feuda, R.; Dohrmann, M.; Pett, W.; Philippe, H.; Rota-Stabelli, O.; Lartillot, N.; Wörheide, G.; Pisani, D. Improved Modeling of Compositional Heterogeneity Supports Sponges as Sister to All Other Animals. Curr. Biol. 2017, 27, 3864–3870.e4. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T.; Saito, T.; Tsunamoto, Y.; Koseki, J.; Ye, B.; Do, V.T.; Miura, O.; Suyama, Y.; Chiba, S. Enigmatic Incongruence between MtDNA and NDNA Revealed by Multi-Locus Phylogenomic Analyses in Freshwater Snails. Sci. Rep. 2019, 9, 6223. [Google Scholar] [CrossRef]
- Near, T.J.; Eytan, R.I.; Dornburg, A.; Kuhn, K.L.; Moore, J.A.; Davis, M.P.; Wainwright, P.C.; Friedman, M.; Smith, W.L. Resolution of Ray-Finned Fish Phylogeny and Timing of Diversification. Proc. Natl. Acad. Sci. USA 2012, 109, 13698–13703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Peng, R.; Kuro-O, M.; Zeng, X. Exploring Patterns and Extent of Bias in Estimating Divergence Time from Mitochondrial DNA Sequence Data in a Particular Lineage: A Case Study of Salamanders (Order Caudata). Mol. Biol. Evol. 2011, 28, 2521–2535. [Google Scholar] [CrossRef]
- Wahlberg, N.; Weingartner, E.; Warren, A.D.; Nylin, S. Timing Major Conflict between Mitochondrial and Nuclear Genes in Species Relationships of Polygonia Butterflies (Nymphalidae: Nymphalini). BMC Evol. Biol. 2009, 9, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reidenbach, K.R.; Cook, S.; Bertone, M.A.; Harbach, R.E.; Wiegmann, B.M.; Besansky, N.J. Phylogenetic Analysis and Temporal Diversification of Mosquitoes (Diptera: Culicidae) Based on Nuclear Genes and Morphology. BMC Evol. Biol. 2009, 9, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, A.F.; Machado, L.C.; de Paula, M.B.; da Silva Pessoa Vieira, C.J.; de Morais Bronzoni, R.V.; de Melo Santos, M.A.V.; Wallau, G.L. Culicidae Evolutionary History Focusing on the Culicinae Subfamily Based on Mitochondrial Phylogenomics. Sci. Rep. 2020, 10, 18823. [Google Scholar] [CrossRef]
- Reisen, W.K. Update on Journal Policy of Aedine Mosquito Genera and Subgenera. J. Med. Entomol. 2016, 53, 249. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple Alignment of Nucleotide Sequences Guided by Amino Acid Translations. Nucleic Acids Res. 2010, 38, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2018, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kück, P.; Meusemann, K. FASconCAT: Convenient Handling of Data Matrices. Mol. Phylogenet. Evol. 2010, 56, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian Software Package for Phylogenetic Reconstruction and Molecular Dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2019, 15, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Benton, M.J.; Donoghue, P.C.J. Paleontological Evidence to Date the Tree of Life. Mol. Biol. Evol. 2007, 24, 26–53. [Google Scholar] [CrossRef]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics Resolves the Timing and Pattern of Insect Evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Borkent, A.; Grimaldi, D.A. The Earliest Fossil Mosquito (Diptera: Culicidae), in Mid-Cretaceous Burmese Amber. Ann. Entomol. Soc. Am. 2004, 97, 882–888. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lartillot, N.; Philippe, H. A Bayesian Mixture Model for Across-Site Heterogeneities in the Amino-Acid Replacement Process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Rota-Stabelli, O.; Daley, A.C.; Pisani, D. Molecular Timetrees Reveal a Cambrian Colonization of Land and a New Scenario for Ecdysozoan Evolution. Curr. Biol. 2013, 23, 392–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, 699–710. [Google Scholar] [CrossRef]
- Richards, S.; Murali, S.C. Best Practices in Insect Genome Sequencing: What Works and What Doesn’t. Curr. Opin. Insect Sci. 2015, 7, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Kooi, C.J.; Ollerton, J. The Origins of Flowering Plants and Pollinators. Science 2020, 368, 1306–1308. [Google Scholar] [CrossRef]
- Rota-Stabelli, O.; Telford, M.J. A Multi Criterion Approach for the Selection of Optimal Outgroups in Phylogeny: Recovering Some Support for Mandibulata over Myriochelata Using Mitogenomics. Mol. Phylogenet. Evol. 2008, 48, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Braband, A.; Middendorf, M.; Misof, B.; Rota-Stabelli, O.; Stadler, P.F. Bioinformatics Methods for the Comparative Analysis of Metazoan Mitochondrial Genome Sequences. Mol. Phylogenet. Evol. 2013, 69, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Reeves, L.E.; Holderman, C.J.; Blosser, E.M.; Gillett-Kaufman, J.L.; Kawahara, A.Y.; Kaufman, P.E.; Burkett-Cadena, N.D. Identification of Uranotaenia Sapphirina as a Specialist of Annelids Broadens Known Mosquito Host Use Patterns. Commun. Biol. 2018, 1, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Rota-Stabelli, O.; Kayal, E.; Gleeson, D.; Daub, J.; Boore, J.L.; Telford, M.J.; Pisani, D.; Blaxter, M.; Lavrov, D.V. Ecdysozoan Mitogenomics: Evidence for a Common Origin of the Legged Invertebrates, the Panarthropoda. Genome Biol. Evol. 2010, 2, 425–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulou, A.; Anastasiou, I.; Vogler, A.P. Revisiting the Insect Mitochondrial Molecular Clock: The Mid-Aegean Trench Calibration. Mol. Biol. Evol. 2010, 27, 1659–1672. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.Y.W.; Lanfear, R.; Bromham, L.; Phillips, M.J.; Soubrier, J.; Rodrigo, A.G.; Cooper, A. Time-Dependent Rates of Molecular Evolution. Mol. Ecol. 2011, 20, 3087–3101. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.W.; Lo, N. The Insect Molecular Clock. Aust. J. Entomol. 2013, 52, 101–105. [Google Scholar] [CrossRef]
- Guidetti, R.; McInnes, S.J.; Cesari, M.; Rebecchi, L.; Rota-Stabelli, O. Evolutionary Scenarios for the Origin of an Antarctic Tardigrade Species Based on Molecular Clock Analyses and Biogeographic Data. Contrib. Zool. 2017, 86, 97–110. [Google Scholar] [CrossRef] [Green Version]
- Andújar, C.; Serrano, J.; Gámez-Zurita, J. Winding up the Molecular Clock in the Genus Carabus (Coleoptera: Carabidae): Assessment of Methodological Decisions on Rate and Node Age Estimation. BMC Evol. Biol. 2012, 12, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thawornwattana, Y.; Dalquen, D.; Yang, Z. Coalescent Analysis of Phylogenomic Data Confidently Resolves the Species Relationships in the Anopheles Gambiae Species Complex. Mol. Biol. Evol. 2018, 35, 2512–2527. [Google Scholar] [CrossRef]
- Foster, P.G.; de Oliveira, T.M.P.; Bergo, E.S.; Conn, J.E.; Sant’Ana, D.C.; Nagaki, S.S.; Nihei, S.; Lamas, C.E.; González, C.; Moreira, C.C.; et al. Phylogeny of Anophelinae Using Mitochondrial Protein Coding Genes. R. Soc. Open Sci. 2017, 4, 170758. [Google Scholar] [CrossRef] [Green Version]
- Palatini, U.; Masri, R.A.; Cosme, L.V.; Koren, S.; Thibaud-Nissen, F.; Biedler, J.K.; Krsticevic, F.; Johnston, J.S.; Halbach, R.; Crawford, J.E.; et al. Improved Reference Genome of the Arboviral Vector Aedes Albopictus. Genome Biol. 2020, 21, 215. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clock Model | Substitution Model | Tree Prior | logLikelihood | AICm | Harmonic Mean | PS/SS | Culicidae | Aedini |
|---|---|---|---|---|---|---|---|---|
| Strict | GTR | Yule | 54,908.6 | 109,892.3 | −54,926.7 | 4 | 156 (114–204) | 90 (75–104) |
| Relaxed (LogN) | HKY | Yule | 54,624.8 | 109,467.9 | −54,666.3 | 5 | 166 (119–215) | 96 (68–125) |
| GTR | Yule | 54,362.7 | 109,108.1 | −54,424.3 | 1 | 180 (137–228) | 113 (83–143) | |
| Birth Death | 54,363.6 | 109,115 | −54,414.9 | 1 | 180 (135–227) | 112 (82–142) | ||
| Coalescent Constant | 54,370.2 | 109,208.7 | −54,415.7 | 3 | 173 (123–225) | 100 (66–132) |
| Node | Taxonomic Level | Concatenated Dataset Figure 3 | No Outgroup (Concatenated) | Nuclear Data Figure 4A | Mitochondrial Data Figure 4B | Others: Reidenbach09; Soghigian17 *; Da Silva 20 #; Chen 15 ^ |
|---|---|---|---|---|---|---|
| a | Diptera | 257 (223–294) | 261 (225–296) | 258 (224–293) | 260 (239–295) ^ | |
| b | Culicidae split (Culicinae origin) | 180 (137–228) | 100 (50–185) | 178 (113–245) | 182 (143–223) | 216 (229–192) 182 # 218 (181–260) ^ |
| c | Culicinae split | 146 (108–182) | 92 (41–139) | 139 (92–194) | 150(118–184) | 204 (226–172) 130 # 179 (148–217) ^ |
| d | 137 (103–173) | 86 (38–127) | 123 (79–171) | 135 (104–164) | ||
| e | Aedini split (Aedes origin) | 113 (83–143) | 64 (34–122) | 92 (55–137) | 111 (95–150) | 123 (155–90) 125 * 102 # |
| f | Aedes split (Clades A-B split) | 105 (77–133) | 57 (28–110) | 69 (42–103) | 107 (85–133) | 92 (123–61) 102 * |
| g | Clade A split | 99 (72–126) | 51 (24–100) | 49 (29–76) | 96 (73–118) | |
| h | 83 (59–109) | 50 (22–93) | 36 (20–57) | 92 (71–116) | ||
| i | Stegomya (A. aegypti–A. albopictus) split | 73 (50–96) | 36 (14–70) | 27 (15–45) | 81 (61–102) | 55 * 67 # 71 (44–107) ^ |
| j | A. albopictus–A. flavopictus split | 28 (14–43) | 36 (14–70) | 3.7 (0.1–11.2) | 33 (20–46) | 25 * |
| l | A. koreicus–A. japonicas split | 32 (15–51) | 14 (3–31) | 3.6 (0.2–10.9) | 46 (24–71) | 20 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zadra, N.; Rizzoli, A.; Rota-Stabelli, O. Chronological Incongruences between Mitochondrial and Nuclear Phylogenies of Aedes Mosquitoes. Life 2021, 11, 181. https://doi.org/10.3390/life11030181
Zadra N, Rizzoli A, Rota-Stabelli O. Chronological Incongruences between Mitochondrial and Nuclear Phylogenies of Aedes Mosquitoes. Life. 2021; 11(3):181. https://doi.org/10.3390/life11030181
Chicago/Turabian StyleZadra, Nicola, Annapaola Rizzoli, and Omar Rota-Stabelli. 2021. "Chronological Incongruences between Mitochondrial and Nuclear Phylogenies of Aedes Mosquitoes" Life 11, no. 3: 181. https://doi.org/10.3390/life11030181
APA StyleZadra, N., Rizzoli, A., & Rota-Stabelli, O. (2021). Chronological Incongruences between Mitochondrial and Nuclear Phylogenies of Aedes Mosquitoes. Life, 11(3), 181. https://doi.org/10.3390/life11030181