
The Mitochondrial Genome of a Plant Fungal Pathogen Pseudocercospora fijiensis (Mycosphaerellaceae), Comparative Analysis and Diversification Times of the Sigatoka Disease Complex Using Fossil Calibrated Phylogenies

Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strain, DNA Extraction, and Library Construction and Sequencing
2.2. Sequence Sources, Data Filtering and Assemblies
2.3. Annotation
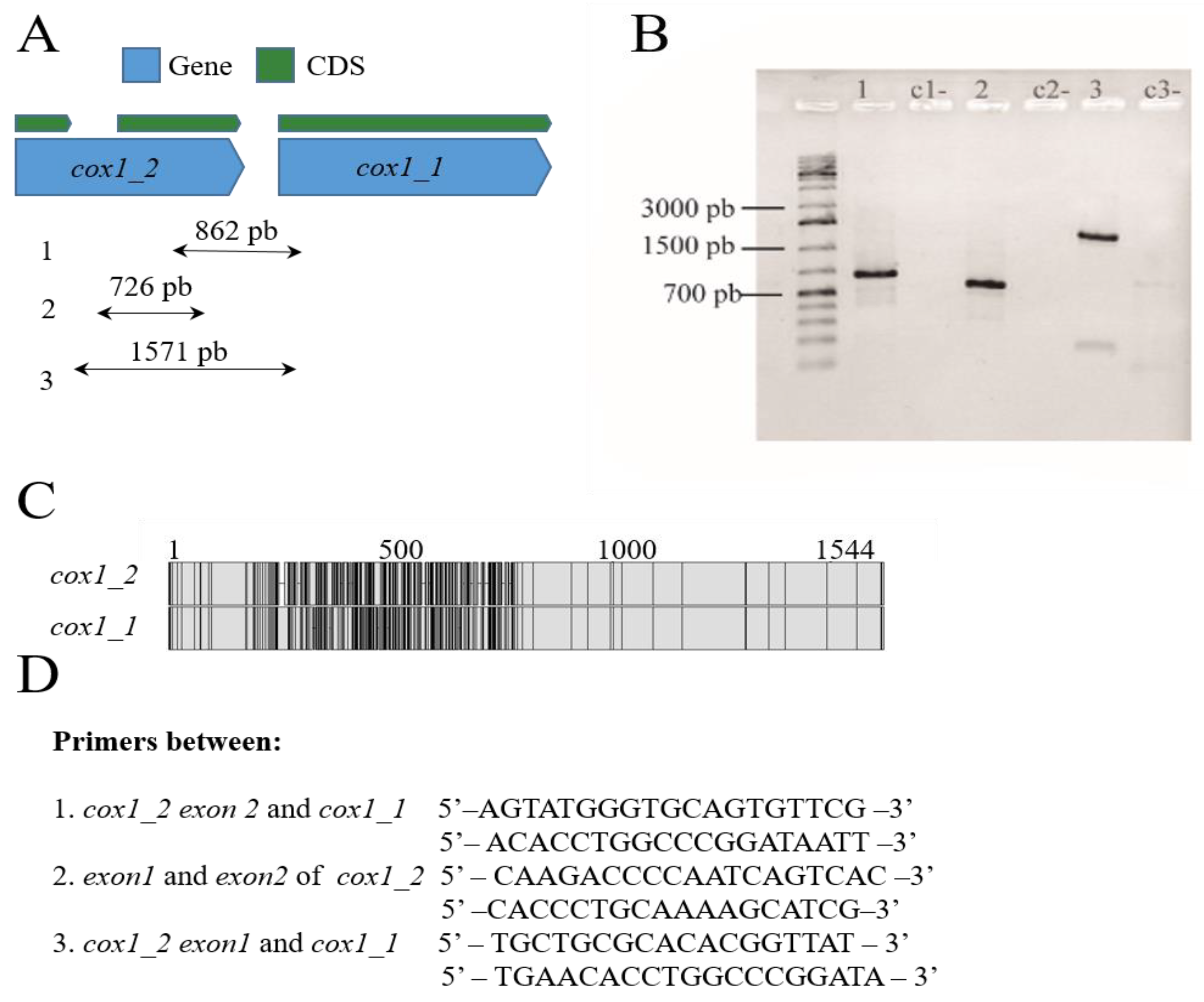
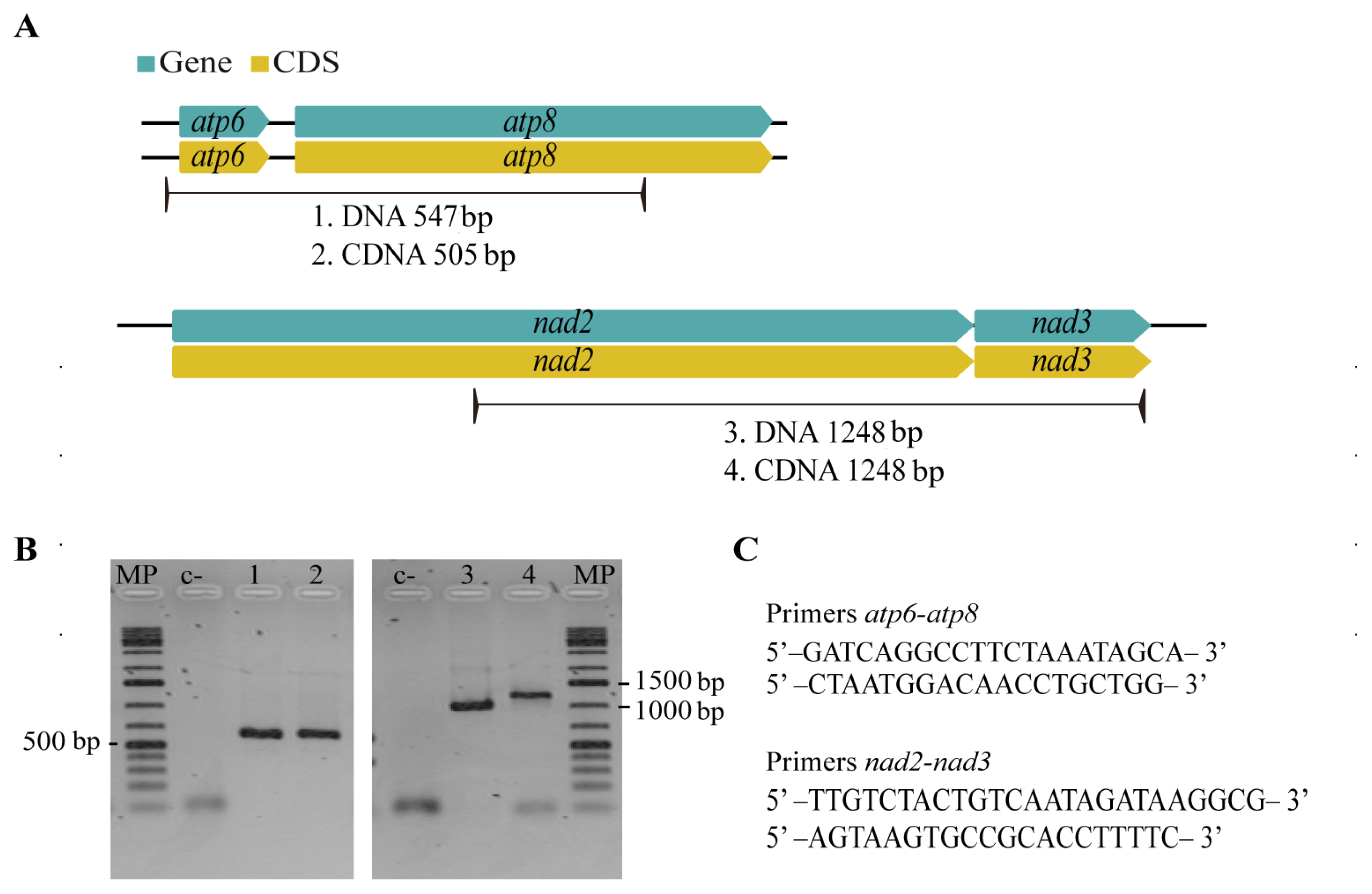
2.4. PCR Amplification of cox1 Gene Copies in P. fijiensis
2.5. Transcriptome de novo Assembly
2.6. RT–PCR Assays for Mitochondrial Gene Pairs of P. fijiensis
2.7. Identification of Repetitive Elements
2.8. Phylogenetic Analysis and Divergence Times Estimates
3. Results
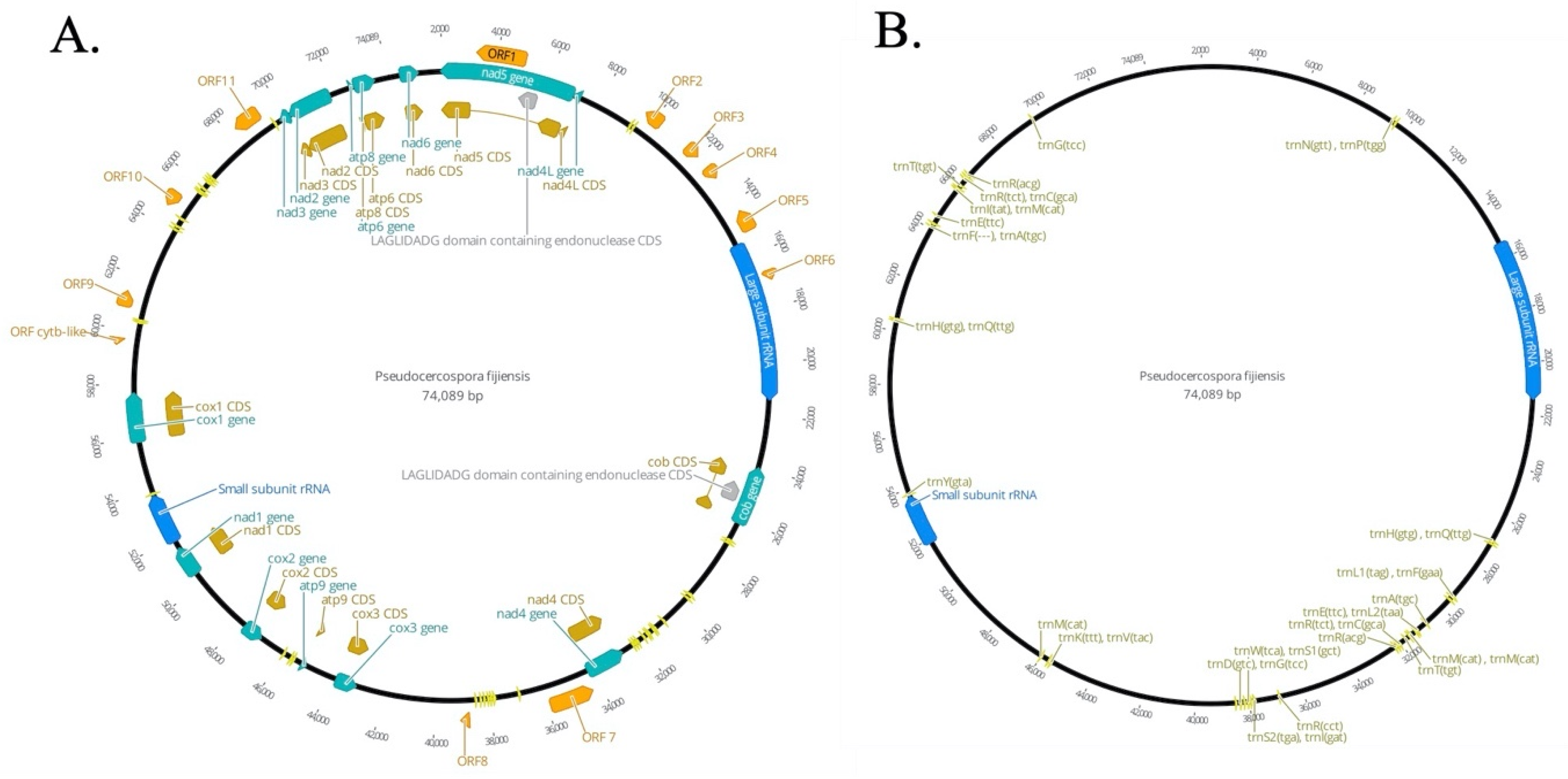
3.1. P. fijiensis Mitochondrial Genome
3.1.1. Presence of Truncated Conserved Mitochondrial Protein-Coding Genes (CMPCGs)
3.1.2. Homing Endonucleases and Introns in the P. fijiensis Mitogenome
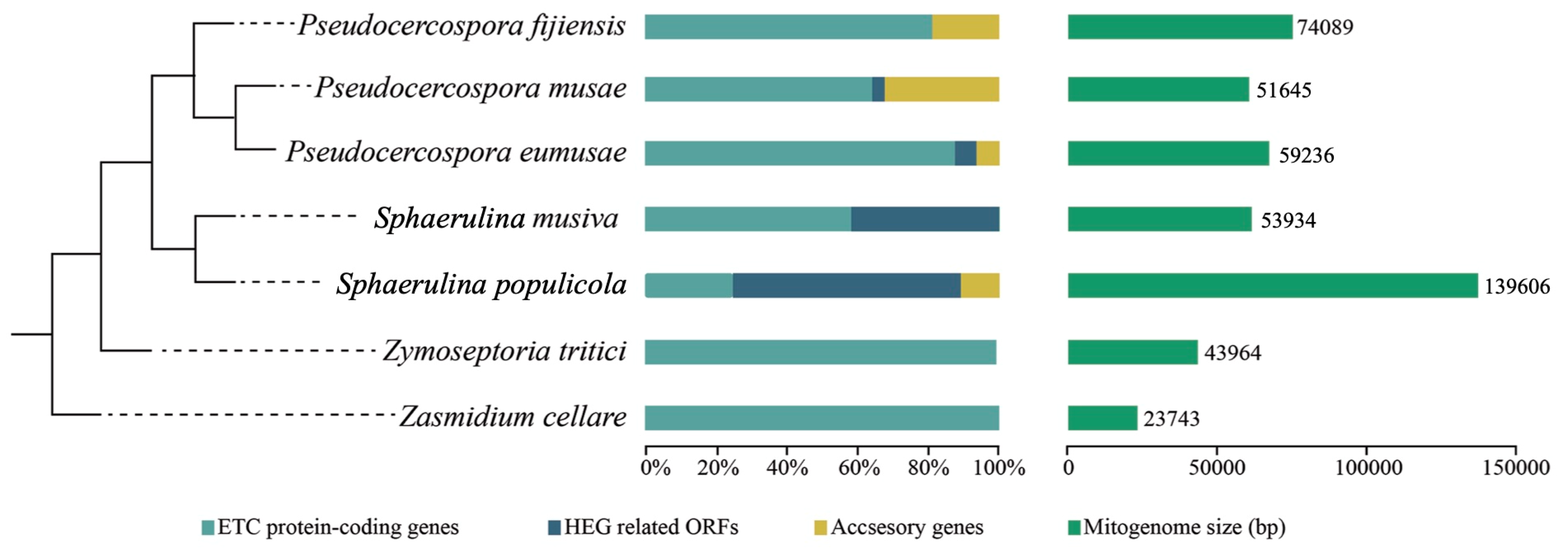
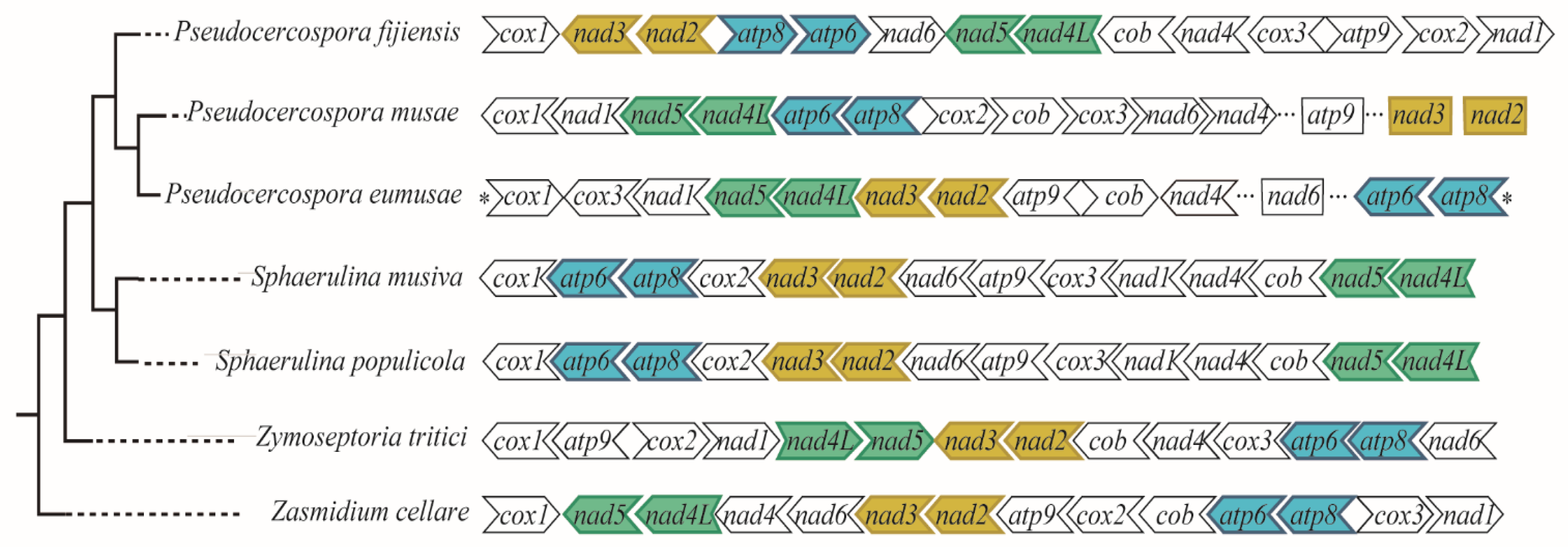
3.2. Comparative Analysis among Mitochondrial Genomes of Selected Mycosphaerellaceae
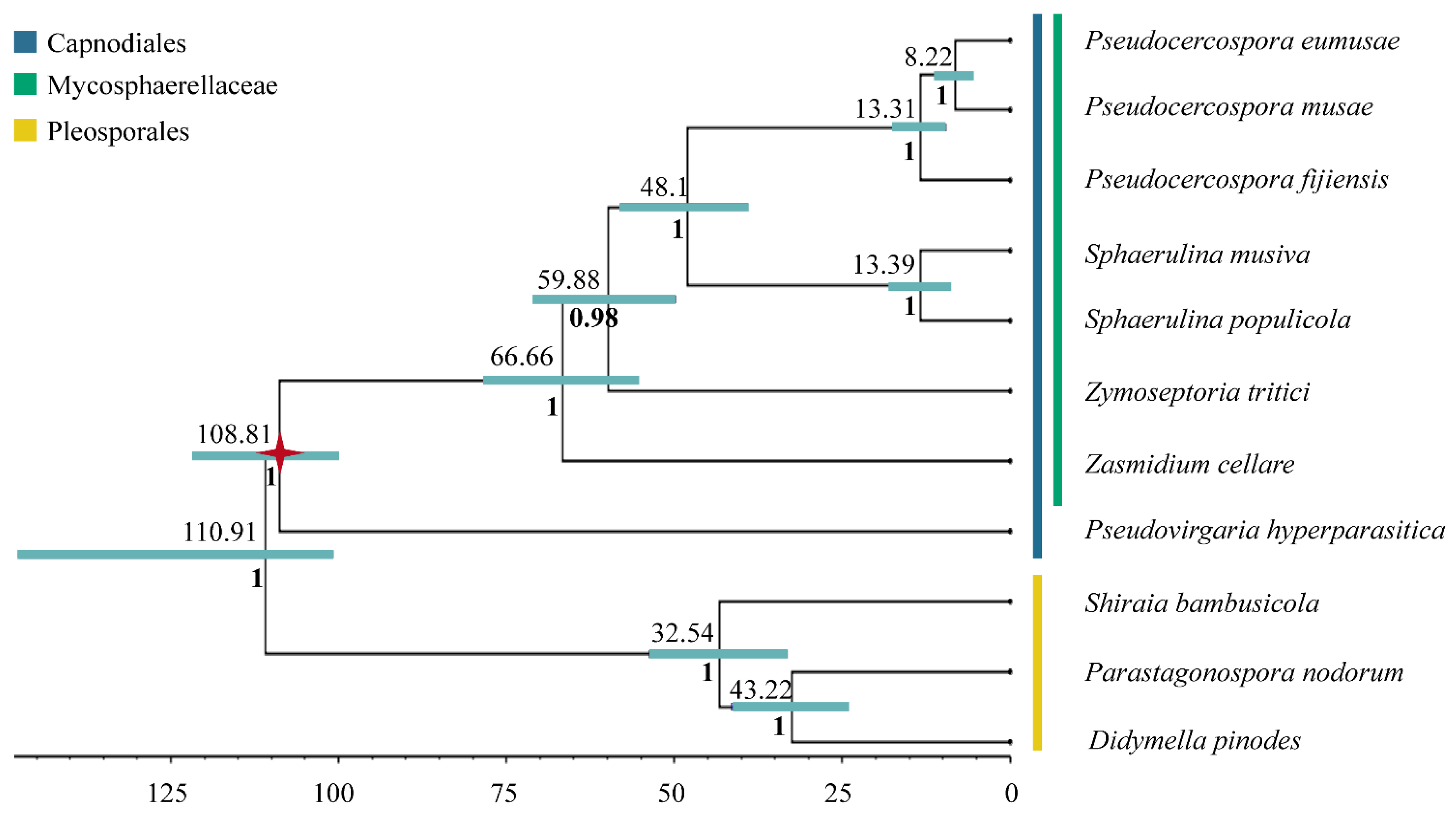
3.3. Inferred Mitochondrial Phylogeny and Diversification Times
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wijayawardene, N.N.; Crous, P.W.; Kirk, P.M.; Hawksworth, D.L.; Boonmee, S.; Braun, U.; Dai, D.Q.; D’souza, M.J.; Diederich, P.; Dissanayake, A.; et al. Naming and outline of Dothideomycetes-2014 including proposals for the protection or suppression of generic names. Fungal. Divers. 2014, 69, 1–55. [Google Scholar] [CrossRef]
- Videira, S.I.R.; Groenewald, J.Z.; Nakashima, C.; Braun, U.; Barreto, R.W.; de Wit, P.J.; Crous, P.W. Mycosphaerellaceae—Chaos or clarity? Stud. Mycol. 2017, 87, 257–421. [Google Scholar] [CrossRef] [PubMed]
- Carlier, J.; Zapater, M.-F.; Lapeyre, F.; Jones, D.R.; Mourichon, X. Septoria Leaf Spot of Banana: A Newly Discovered Disease Caused by Mycosphaerella eumusae (Anamorph Septoria eumusae). Phytopathology 2000, 90, 884–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomma, B.P.; Van Esse, H.P.; Crous, P.W.; de Wit, P.J. Cladosporium fulvum (syn. Passalora fulva), a highly specialized plant pathogen as a model for functional studies on plant pathogenic Mycosphaerellaceae. Mol. Plant Pathol. 2005, 6, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Stukenbrock, E.H.; Banke, S.; Javan-Nikkhah, M.; McDonald, B.A. Origin and Domestication of the Fungal Wheat Pathogen Mycosphaerella graminicola via Sympatric Speciation. Mol. Biol. Evol. 2006, 24, 398–411. [Google Scholar] [CrossRef] [Green Version]
- De Lapeyre, L.; Bellaire, D.; Fouré, E.; Abadie, C.; Carlier, J. Black Leaf Streak Disease is challenging the banana industry. Fruits 2010, 65, 327–342. [Google Scholar] [CrossRef]
- Drenkhan, R.; Adamson, K.; Jürimaa, K.; Hanso, M. Dothistroma septosporum on firs (Abies spp.) in the northern Baltics. For. Pathol. 2014, 44, 250–254. [Google Scholar] [CrossRef]
- Dhillon, B.; Feau, N.; Aerts, A.L.; Beauseigle, S.; Bernier, L.; Copeland, A.; Foster, A.; Navdeep, G.; Henrissat, B.; Herath, P.; et al. Horizontal gene transfer and gene dosage drives adaptation to wood colonization in a tree pathogen. Proc. Natl. Acad. Sci. USA 2015, 112, 3451–3456. [Google Scholar] [CrossRef] [Green Version]
- Arzanlou, M.; Groenewald, J.Z.; Fullerton, R.A.; Abeln, E.C.A.; Carlier, J.; Zapater, M.-F.; Buddenhagen, W.; Viljoen, A.; Crous, P.W. Multiple gene genealogies and phenotypic characters differentiate several novel species of Mycosphaerella and related anamorphs on banana. Pers. Mol. Phylogeny Evol. Fungi 2008, 20, 19–37. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.C.; Salvucci, A.; Crous, P.W.; Stergiopoulos, I. Comparative Genomics of the Sigatoka Disease Complex on Banana Suggests a Link between Parallel Evolutionary Changes in Pseudocercospora fijiensis and Pseudocercospora eumusae and Increased Virulence on the Banana Host. PLoS Genet. 2016, 12, e1005904. [Google Scholar] [CrossRef] [PubMed]
- Ohm, R.A.; Feau, N.; Henrissat, B.; Schoch, C.L.; Horwitz, B.A.; Barry, K.W.; Condon, B.J.; Copeland, A.C.; Dhillon, B.; Glaser, F.; et al. Diverse Lifestyles and Strategies of Plant Pathogenesis Encoded in the Genomes of Eighteen Dothideomycetes Fungi. PLoS Pathog. 2012, 8, e1003037. [Google Scholar] [CrossRef] [Green Version]
- Arango Isaza, R.E.; Diaz-Trujillo, C.; Dhillon, B.; Aerts, A.; Carlier, J.; Crane, C.F.; de Jong, T.V.; de Vries, I.; Dietrich, R.; Farmer, A.D.; et al. Combating a Global Threat to a Clonal Crop: Banana Black Sigatoka Pathogen Pseudocercospora fijiensis (Synonym Mycosphaerella fijiensis) Genomes Reveal Clues for Disease Control. PLoS Genet. 2016, 12, e1005876. [Google Scholar] [CrossRef]
- Churchill, A.C.L. Mycosphaerella fijiensis, the black leaf streak pathogen of banana: Progress towards understanding pathogen biology and detection, disease development, and the challenges of control. Mol. Plant. Pathol. 2011, 12, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Forget, L.; Ustinova, J.; Wang, Z.; Huss, V.A.R.; Franz Lang, B. Hyaloraphidium curvatum: A Linear Mitochondrial Genome, tRNA Editing, and an Evolutionary Link to Lower Fungi. Mol. Biol. Evol. 2002, 19, 310–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Cai, Y.; Zhang, Q.; Chen, L.; Shu, F.; Ma, X.; Bian, Y. The mitochondrial genome of Morchella importuna (272.2 kb) is the largest among fungi and contains numerous introns, mitochondrial non-conserved open reading frames and repetitive sequences. Int. J. Biol. Macromol. 2020, 143, 373–381. [Google Scholar] [CrossRef]
- Hausner, G. Fungal mitochondrial genomes. Fungal Genomics. 2003, 3, 101. [Google Scholar]
- Clark-Walker, G.D. Evolution of Mitochondrial Genomes in Fungi. Int. Rev. Cytol. 1992, 141, 89–127. [Google Scholar] [CrossRef]
- Wolf, K.; Giudice, L.D. The Variable Mitochondrial Genome of Ascomycetes: Organization, Mutational Alterations, and Expression. Adv. Genet. 1988, 25, 185–308. [Google Scholar]
- Paquin, B.; Laforest, M.J.; Forget, L.; Roewer, I.; Wang, Z.; Longcore, J.; Lang, B.F. The fungal mitochondrial genome project: Evolution of fungal mitochondrial genomes and their gene expression. Curr. Genet. 1997, 31, 380–395. [Google Scholar] [CrossRef]
- Aguileta, G.; De Vienne, D.M.; Ross, O.N.; Hood, M.E.; Giraud, T.; Petit, E.; Gabaldón, T. High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 2014, 6, 451–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, S.; Takano, H.; Kuroiwa, T. Sexuality of Mitochondria: Fusion, Recombination, and Plasmids. Int. Rev. Cytol. 1995, 161, 49–110. [Google Scholar] [CrossRef]
- Burger, G.; Gray, M.W.; Lang, B.F. Mitochondrial genomes: Anything goes. Trends Genet. 2003, 19, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Franz Lang, B.; Laforest, M.-J.; Burger, G. Mitochondrial introns: A critical view. Trends Genet. 2007, 23, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Alverson, A.J.; Wei, X.; Rice, D.W.; Stern, D.B.; Barry, K.; Palmer, J.D. Insights into the Evolution of Mitochondrial Genome Size from Complete Sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 2010, 27, 1436–1448. [Google Scholar] [CrossRef] [Green Version]
- Bernt, M.; Braband, A.; Schierwater, B.; Stadler, P.F. Genetic aspects of mitochondrial genome evolution. Mol. Phylogenet. Evol. 2013, 69, 328–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torriani, S.F.F.; Penselin, D.; Knogge, W.; Felder, M.; Taudien, S.; Platzer, M.; McDonald, B.A.; Brunner, P.C. Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal. Genet. Biol. 2014, 62, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Basse, C.W. Mitochondrial inheritance in fungi. Curr. Opin. Microbiol. 2010, 13, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.J.F. Mitochondrial inheritance in filamentous fungi. J. Genet. 1996, 75, 403–414. [Google Scholar] [CrossRef]
- Beimforde, C.; Feldberg, K.; Nylinder, S.; Rikkinen, J.; Tuovila, H.; Dörfelt, H.; Gube, M.; Jackson, D.J.; Reitner, J.; Seyfullah, L.J.; et al. Estimating the Phanerozoic history of the Ascomycota lineages: Combining fossil and molecular data. Mol. Phylogenet. Evol. 2014, 78, 386–398. [Google Scholar] [CrossRef]
- Torriani, S.F.F.; Goodwin, S.B.; Kema, G.H.J.; Pangilinan, J.L.; McDonald, B.A. Intraspecific comparison and annotation of two complete mitochondrial genome sequences from the plant pathogenic fungus Mycosphaerella graminicola. Fungal. Genet. Biol. 2008, 45, 628–637. [Google Scholar] [CrossRef]
- Goodwin, S.B.; Mccorison, C.B.; Cavaletto, J.R.; Culley, D.E.; Labutti, K.; Baker, S.E.; Grigoriev, I.V. The mitochondrial genome of the ethanol-metabolizing, wine cellar mold Zasmidium cellare is the smallest for a filamentous ascomycete. Fungal. Biol. 2016, 120, 961–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johanson, A.; Jeger, M.J. Use of PCR for detection of Mycosphaerella fijiensis and M. musicola, the causal agents of Sigatoka leaf spots in banana and plantain. Mycol. Res. 1993, 97, 670–674. [Google Scholar] [CrossRef]
- Cañas-Gutiérrez, G.P.; Angarita-Velásquez, M.J.; Restrepo-Flórez, J.M.; Rodríguez, P.; Moreno, C.X.; Arango, R. Analysis of the CYP51 gene and encoded protein in propiconazole-resistant isolates of Mycosphaerella fijiensis. Pest. Manag. Sci. 2009, 65, 892–899. [Google Scholar] [CrossRef]
- Benson, D.A.; Boguski, M.S.; Lipman, D.J.; Ostell, J.; Ouellette, B.F.F.; Rapp, B.A.; Wheeler, D.L. GenBank. Nucleic. Acids. Res. 1999, 27, 12–17. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic. Acids. Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.; Otillar, R.; Riley, R.; Salamov, A.; Zhao, X.; Korzeniewski, F.; et al. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids. Res. 2014, 42, D699–D704. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 March 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadalin, F.; Vezzi, F.; Policriti, A. GapFiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinform. 2012, 13 (Suppl. 14), S8. [Google Scholar] [CrossRef] [Green Version]
- Hunt, M.; Kikuchi, T.; Sanders, M.; Newbold, C.; Berriman, M.; Otto, T.D. REAPR: A universal tool for genome assembly evaluation. Genome Biol. 2013, 14, R47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hane, J.K.; Lowe, R.G.T.; Solomon, P.S.; Tan, K.-C.; Schoch, C.L.; Spatafora, J.W.; Crous, P.W.; Kodira, C.; Birren, B.W.; Galagan, J.E.; et al. Dothideomycete plant interactions illuminated by genome sequencing and EST analysis of the wheat pathogen Stagonospora nodorum. Plant Cell 2007, 19, 3347–3368. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.-Y.; Li, T.; Chen, S.; Fan, L.; Gao, J.; Hou, C.-L. Characterization and Phylogenetic Analysis of the Mitochondrial Genome of Shiraia bambusicola Reveals Special Features in the Order of Pleosporales. PLoS ONE 2015, 10, e0116466. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Okorski, A.; Pszczółkowska, A.; Jastrzębski, J.P.; Paukszto, Ł.; Okorska, S. The complete mitogenome of Mycosphaerella pinodes (Ascomycota, Mycosphaerellaceae). Mitochondrial DNA Part B 2016, 1, 48–49. [Google Scholar] [CrossRef] [Green Version]
- Braun, U.; Crous, P.W.; Groenewald, J.Z.; Scheuer, C. Pseudovirgaria, a fungicolous hyphomycete genus. IMA Fungus 2011, 2, 65–69. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, G. Estimating the Dimension of a Model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, 88. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.R.; Beimforde, C.; Seyfullah, L.J.; Wege, S.-E.; Dörfelt, H.; Girard, V.; Grabenhorst, H.; Gube, M.; Heinrichs, J.; Nel, A.; et al. Amber fossils of sooty moulds. Rev. Palaeobot Palynol. 2014, 200, 53–64. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A. Tracer: MCMC Trace Analysis Tool, Version 1.5.; University of Oxford: Oxford, UK, 2009; Available online: http://tree.bio.ed.ac.uk/software/tracer/ (accessed on 1 March 2021).
- Rambaut, A. FigTree: Tree Figure Drawing Tool, Version 1.3.1. 2006. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 1 March 2021).
- Megarioti, A.H.; Kouvelis, V.N. The Coevolution of Fungal Mitochondrial Introns and Their Homing Endonucleases (GIY-YIG and LAGLIDADG). Genome. Biol. Evol. 2020, 12, 1337–1354. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.J. Natural plasmids of filamentous fungi. Microbiol. Rev. 1995, 59, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Cahan, P.; Kennell, J.C. Identification and distribution of sequences having similarity to mitochondrial plasmids in mitochondrial genomes of filamentous fungi. Mol. Genet. Genom. 2005, 273, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Jelen, V.; de Jonge, R.; Van de Peer, Y.; Javornik, B.; Jakše, J. Complete mitochondrial genome of the Verticillium-wilt causing plant pathogen Verticillium nonalfalfae. PLoS ONE 2016, 11, e0148525. [Google Scholar] [CrossRef] [Green Version]
- Salvatori, R. Organization and Regulation of Mitochondrial Gene Expression; Stockholm University: Stockholm, Sweden, 2020; Available online: https://www.diva-portal.org/smash/get/diva2:1416629/FULLTEXT01.pdf?fbclid=IwAR1Ncc4BTQ26Ygmh_aPBdwvK6HDsivbSSp44OIHnBdMdsSYEzCTqT6P0Bwo (accessed on 1 July 2017).
- Joardar, V.; Abrams, N.F.; Hostetler, J.; Paukstelis, P.J.; Pakala, S.; Pakala, S.B.; Zafar, N.; Abolude, O.O.; Payne, J.; Andrianopoulos, A.; et al. Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genom. 2012, 13, 698. [Google Scholar] [CrossRef]
- Mardanov, A.V.; Beletsky, A.V.; Kadnikov, V.V.; Ignatov, A.N.; Ravin, N.V. The 203 kbp mitochondrial genome of the phytopathogenic fungus Sclerotinia borealis reveals multiple invasions of introns and genomic duplications. PLoS ONE 2014, 9, e107536. [Google Scholar] [CrossRef]
- Dassa, B.; London, N.; Stoddard, B.L.; Schueler-Furman, O.; Pietrokovski, S. Fractured genes: A novel genomic arrangement involving new split inteins and a new homing endonuclease family. Nucleic Acids Res. 2009, 37, 2560–2573. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gueidan, C.; Ruibal, C.; de Hoog, G.S.; Schneider, H. Rock-inhabiting fungi originated during periods of dry climate in the late Devonian and middle Triassic. Fungal. Biol. 2011, 115, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Christelová, P.; Valárik, M.; Eva, H.; Langhe, E.D.; Dole, J. A multi gene sequence-based phylogeny of the Musaceae (banana) family. BMC Evol. Biol. 2011, 11, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Start Position | Stop Position | Length (bp) | Direction | Start Codon | Stop Codon | GC Contents | Product |
|---|---|---|---|---|---|---|---|---|
| nad6 | 435 | 1109 | 675 | forward | ATA | TAA | 23% | subunit 6 of NADH dehydrogenase |
| nad5 | 1922 | 7012 | 5091 | reverse | ATA | TAA | 24.8 | NADH dehydrogenase subunit 5 |
| ORF1 | 3253 | 5001 | 1749 | reverse | ATG | TAA | 22.8 | hypothetical protein |
| LAGLIDADG | 5042 | 5722 | 681 | reverse | TTA | TAA | 24.4 | LAGLIDADG domain containing endonuclease |
| nad4L | 7009 | 7278 | 270 | reverse | TTA | TAA | 23.7 | NADH dehydrogenase subunit 4L |
| trnN(gtt) | 9370 | 9440 | 71 | reverse | - | - | - | tRNA-Asn |
| trnP(tgg) | 9480 | 9552 | 73 | reverse | - | - | - | |
| ORF2 | 9600 | 10271 | 672 | reverse | ATG | TAA | 29.9 | hypothetical protein |
| ORF3 | 11452 | 11973 | 522 | forward | TTA | TAG | 26.2 | hypothetical protein |
| ORF4 | 12481 | 12969 | 489 | forward | ATG | TAA | 33.5 | hypothetical protein |
| ORF5 | 14399 | 15151 | 753 | reverse | ATA | TAA | - | hypothetical protein |
| Large subunit rRNA | 15433 | 21413 | 5981 | forward | - | - | - | large subunit ribosomal RNA |
| ORF6 | 16699 | 17037 | 339 | forward | ATT | TAA | 14.7 | hypothetical protein |
| cob | 23991 | 26209 | 2219 | reverse | TTA | TAA | 29.3 | cytochrome b |
| LAGLIDADG | 24749 | 25492 | 744 | reverse | - | - | - | LAGLIDADG domain containing endonuclease |
| trnH(gtg) | 26876 | 26948 | 73 | reverse | - | - | - | tRNA-His |
| trnQ(ttg) | 26974 | 27046 | 73 | reverse | - | - | - | tRNA-Gln |
| trnL1(tag) | 29371 | 29454 | 84 | reverse | - | - | - | tRNA-Leu |
| trnF(gaa) | 29531 | 29603 | 73 | reverse | - | - | - | tRNA-Phe |
| trnA(tgc) | 30770 | 30841 | 72 | reverse | - | - | - | tRNA-Ala |
| trnE(ttc) | 31173 | 31244 | 72 | reverse | - | - | - | tRNA-Glu |
| trnL2(taa) | 31253 | 31335 | 83 | reverse | - | - | - | tRNA-Leu |
| trnM(cat) | 31478 | 31550 | 73 | reverse | - | - | - | tRNA-Met |
| trnM(cat) | 31555 | 31625 | 71 | reverse | - | - | - | tRNA-Met |
| trnT(tgt) | 31714 | 31784 | 71 | reverse | - | - | - | tRNA-Thr |
| trnR(tct) | 32001 | 32071 | 71 | reverse | - | - | - | tRNA-Arg |
| trnC(gca) | 32105 | 32175 | 71 | reverse | - | - | - | tRNA-Cys |
| trnR(acg) | 32212 | 32290 | 79 | reverse | - | - | - | tRNA-Arg |
| nad4 | 32730 | 34220 | 1491 | reverse | ATA | TAA | 25.4 | NADH dehydrogenase subunit 4 |
| ORF7 | 34319 | 35902 | 1584 | reverse | ATG | TAA | 22.9 | hypothetical protein |
| trnR(cct) | 36840 | 36910 | 71 | reverse | - | - | - | tRNA-Arg |
| trnS2(tga) | 37775 | 37862 | 88 | reverse | - | - | - | tRNA-Ser |
| trnI(gat) | 37911 | 37982 | 72 | reverse | - | - | - | tRNA-Ile |
| trnW(tca) | 38019 | 38090 | 72 | reverse | - | - | - | tRNA-Trp |
| trnS1(gct) | 38154 | 38234 | 81 | reverse | - | - | - | tRNA-Ser |
| trnD(gtc) | 38279 | 38351 | 73 | reverse | - | - | - | tRNA-Asp |
| trnG(tcc) | 38481 | 38551 | 71 | reverse | - | - | - | tRNA-Gly |
| ORF8 | 38798 | 39094 | 297 | forward | ATG | TAG | 19.5 | hypothetical protein |
| cox3 | 43007 | 43816 | 810 | reverse | ATG | TAA | 31.7 | Cytochrome c oxidase subunit III |
| atp9 | 44999 | 45223 | 225 | reverse | ATG | ATG | 40.9 | ATP Synthase Membrane Subunit 9 |
| trnK(ttt) | 45506 | 45578 | 73 | reverse | - | - | - | tRNA-Lys |
| trnV(tac) | 45607 | 45679 | 73 | reverse | - | - | - | tRNA-Val |
| trnM(cat) | 45915 | 45987 | 73 | reverse | - | - | - | tRNA-Met |
| cox2 | 46989 | 47738 | 750 | reverse | TAA | TAA | 29.9 | cytochrome c oxidase subunit II |
| nad1 | 50440 | 51600 | 1161 | forward | ATG | TAA | 30 | NADH dehydrogenase subunit 1 |
| Small subunit rRNA | 51849 | 53773 | 1925 | forward | - | - | - | small subunit ribosomal RNA |
| trnY(gta) | 53797 | 53881 | 85 | forward | - | - | - | tRNA-Tyr |
| cox1 | 55837 | 57708 | 1872 | forward | TTA | TAG | 32.2 | cytochrome c oxidase subunit I |
| ORF cytb-like | 59442 | 59672 | 231 | reverse | TTA | TAA | 22.5 | cytb-like ORF |
| trnH(gtg) | 60291 | 60363 | 73 | reverse | - | - | - | tRNA-His |
| trnQ(ttg) | 60389 | 60461 | 73 | reverse | - | - | - | tRNA-Gln |
| ORF9 | 60802 | 61359 | 558 | forward | ATG | TAA | 35.1 | hypothetical protein |
| trnF(---) | 64038 | 64111 | 74 | reverse | - | - | - | tRNA-Phe |
| trnA(tgc) | 64155 | 64226 | 72 | reverse | - | - | - | tRNA-Ala |
| trnE(ttc) | 64498 | 64569 | 72 | reverse | - | - | - | tRNA-Glu |
| ORF10 | 64714 | 65247 | 534 | reverse | ATA | TAG | 27 | hypothetical protein |
| trnI(tat) | 65672 | 65744 | 73 | reverse | - | - | - | tRNA-Ile |
| trnM(cat) | 65749 | 65819 | 71 | reverse | - | - | - | tRNA-Met |
| trnT(tgt) | 65908 | 65978 | 71 | reverse | - | - | - | tRNA-Thr |
| trnR(tct) | 66182 | 66252 | 71 | reverse | - | - | - | tRNA-Arg |
| trnC(gca) | 66286 | 66356 | 71 | reverse | - | - | - | tRNA-Cys |
| trnR(acg) | 66393 | 66465 | 73 | reverse | - | - | - | tRNA-Arg |
| ORF11 | 68278 | 69276 | 999 | reverse | ATG | TAA | 21.5 | hypothetical protein |
| trnG(tcc) | 69523 | 69593 | 71 | forward | - | - | - | tRNA-Gly |
| nad3 | 69768 | 70139 | 372 | reverse | ATG | TAA | 27.2 | NADH dehydrogenase subunit 3 |
| nad2 | 70140 | 71819 | 1680 | reverse | ATG | TAA | 24.5 | NADH dehydrogenase subunit 2 |
| atp8 | 72609 | 72758 | 150 | forward | ATT | TAA | 24 | ATP Synthase Subunit 8 |
| atp6 | 72801 | 73589 | 789 | forward | ATA | TAA | 27.8 | ATP Synthase Subunit 6 |
| Item | P. musae | P. eumusae | P. fijiensis | S. musiva | S. populicola | Z. cellare | Z. tritici |
|---|---|---|---|---|---|---|---|
| Genome size (bp) | 51,645 | 59,236 | 74,089 | 53,234 | 139,606 | 23,743 | 43,964 |
| GC content (%) | 27.80 | 27 | 19.2 | 24.5 | 31.70 | 27.80 | 31.90 |
| No. of introns | 1 | 1 | 2 | 6 | 28 | 0 | 0 |
| No. of standard Protein Coding Genes (CMPCGs) | 14 | 14 | 14 | 14 | 14 | 14 | 14 |
| No. of rRNAs | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| No. of tRNAs | 24 | 25 | 38 | 29 | 29 | 25 | 27 |
| Genic regions (%) | 63.25 | 40.73 | 65.55 | 65.06 | 98.25 | 79.39 | 67.79 |
| Intergenic regions (%) | 36.75 | 59.27 | 46.45 | 34.94 | 1.75 | 20.51 | 32.21 |
| Number of GIY-YIG intragenic regions | 0 | 0 | 0 | 1 | 5 | 0 | 0 |
| Number of GIY-YIG intergenic regions | 1 | 1 | 0 | 0 | 2 | 0 | 0 |
| Number of LAGLIDADG intragenic regions | 0 | 0 | 0 | 3 | 18 | 0 | 0 |
| Number of LAGLIDADG intergenic regions | 0 | 0 | 2 | 1 | 3 | 0 | 0 |
| Number of Repetitive Sequences | 32 | 43 | 29 | 8 | 20 | 2 | 0 |
| Present Study | Chang et al. 2016 | ||
|---|---|---|---|
| Molecular Clock | Relaxed-Log normal | Strict clock | Strict clock |
| Type of Data | 13 mitochondrial protein-coding genes | 13 mitochondrial protein-coding genes | 46 nuclear single-copy genes |
| Divergence Time estimation method | Bayesian analysis in BEAST2 v2.5.1 | Bayesian analysis in BEAST2 v2.5.1 | Penalized likelihood analysis in the program r8s v1.7 |
| Fossil calibration | Capnodiales-Metacapnodiaceae Fossil | Capnodiales- Metacapnodiaceae Fossil | Dothideomycetes crown group |
| Capnodiales | 108.81 (100.17–121.35 MYA, 95% HPD) | 151.96 (101.04–232.179 MYA, 95% HPD) | 234.2–180.2 MYA |
| Mycosphaerellaceae | 66.66 (55.47–78.27 MYA, 95% HPD) | 97.24 (64.27–155 MYA, 95% HPD) | 186.7–143.6 MYA |
| Sphaerulina | 13.39 (9.39–17.69 MYA, 95% HPD) | 30.31 (19.69–48.63 MYA, 95% HPD) | near 10 MYA |
| Pseudocercospora + Sphaerulina | 48.1 (39.34–57.86 MYA, 95% HPD) | 84.46 (55.6–134.54 MYA, 95% HPD) | 146.6–112.8 MYA |
| Pseudocercospora | 13.31 MYA (9.49–17.28 MYA, 95% HPD) | 27.66 MYA (17.8–44.27MYA, 95% HPD) | 39.9–30.6 MYA |
| P. eumusae + P. musae | 8.22 MYA (5.6–11.07 MYA, 95% HPD) | 17.87 MYA (11.5–28.71 MYA, 95% HPD) | 22.6–17.4 MYA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arcila-Galvis, J.E.; Arango, R.E.; Torres-Bonilla, J.M.; Arias, T. The Mitochondrial Genome of a Plant Fungal Pathogen Pseudocercospora fijiensis (Mycosphaerellaceae), Comparative Analysis and Diversification Times of the Sigatoka Disease Complex Using Fossil Calibrated Phylogenies. Life 2021, 11, 215. https://doi.org/10.3390/life11030215
Arcila-Galvis JE, Arango RE, Torres-Bonilla JM, Arias T. The Mitochondrial Genome of a Plant Fungal Pathogen Pseudocercospora fijiensis (Mycosphaerellaceae), Comparative Analysis and Diversification Times of the Sigatoka Disease Complex Using Fossil Calibrated Phylogenies. Life. 2021; 11(3):215. https://doi.org/10.3390/life11030215
Chicago/Turabian StyleArcila-Galvis, Juliana E., Rafael E. Arango, Javier M. Torres-Bonilla, and Tatiana Arias. 2021. "The Mitochondrial Genome of a Plant Fungal Pathogen Pseudocercospora fijiensis (Mycosphaerellaceae), Comparative Analysis and Diversification Times of the Sigatoka Disease Complex Using Fossil Calibrated Phylogenies" Life 11, no. 3: 215. https://doi.org/10.3390/life11030215
APA StyleArcila-Galvis, J. E., Arango, R. E., Torres-Bonilla, J. M., & Arias, T. (2021). The Mitochondrial Genome of a Plant Fungal Pathogen Pseudocercospora fijiensis (Mycosphaerellaceae), Comparative Analysis and Diversification Times of the Sigatoka Disease Complex Using Fossil Calibrated Phylogenies. Life, 11(3), 215. https://doi.org/10.3390/life11030215