
SAXS Reveals the Stabilization Effects of Modified Sugars on Model Proteins

 , ,
, ,  ,
,  and
and
Abstract
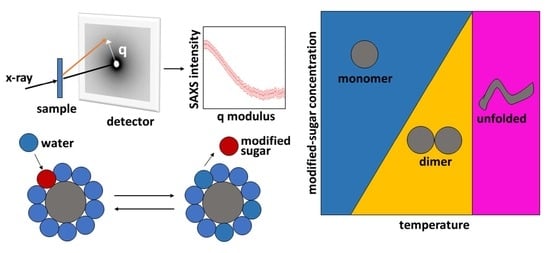
:
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. SAXS Experiments
2.3. SAXS Data Analysis
2.3.1. Multimeric Equilibrium Processes in Binary Solvents
2.3.2. Determination of SLDs
2.3.3. Effective Protein–Protein Structure Factor
2.3.4. Global-Fit of SAXS Data
2.3.5. Myoglobin
2.3.6. Insulin
3. Results
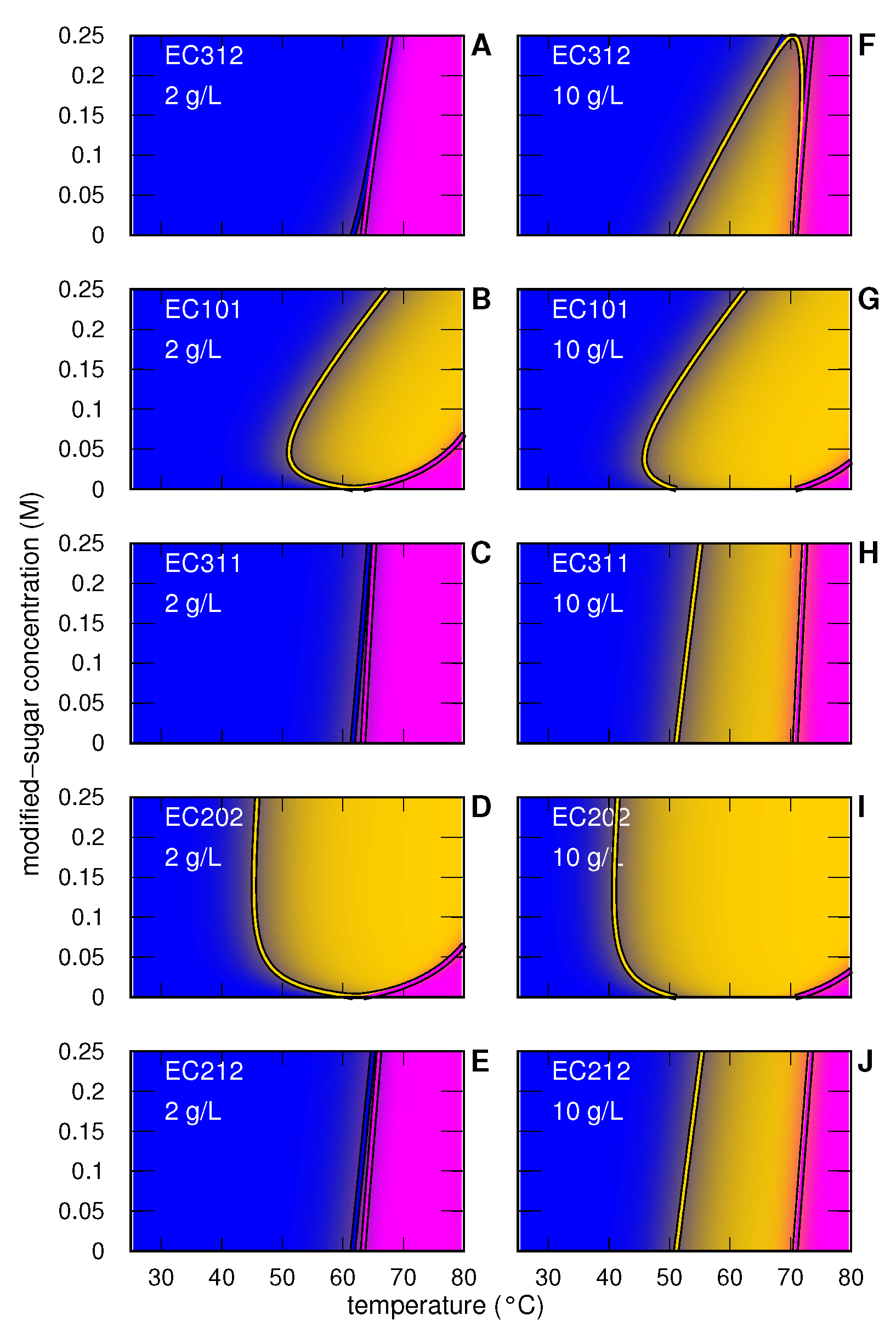
3.1. Myoglobin
3.1.1. Myoglobin without and with Modified Sugar
3.1.2. Myoglobin with EC312
3.1.3. Myoglobin with EC101
3.1.4. Myoglobin with EC311
3.1.5. Myoglobin with EC202
3.1.6. Myoglobin with EC212
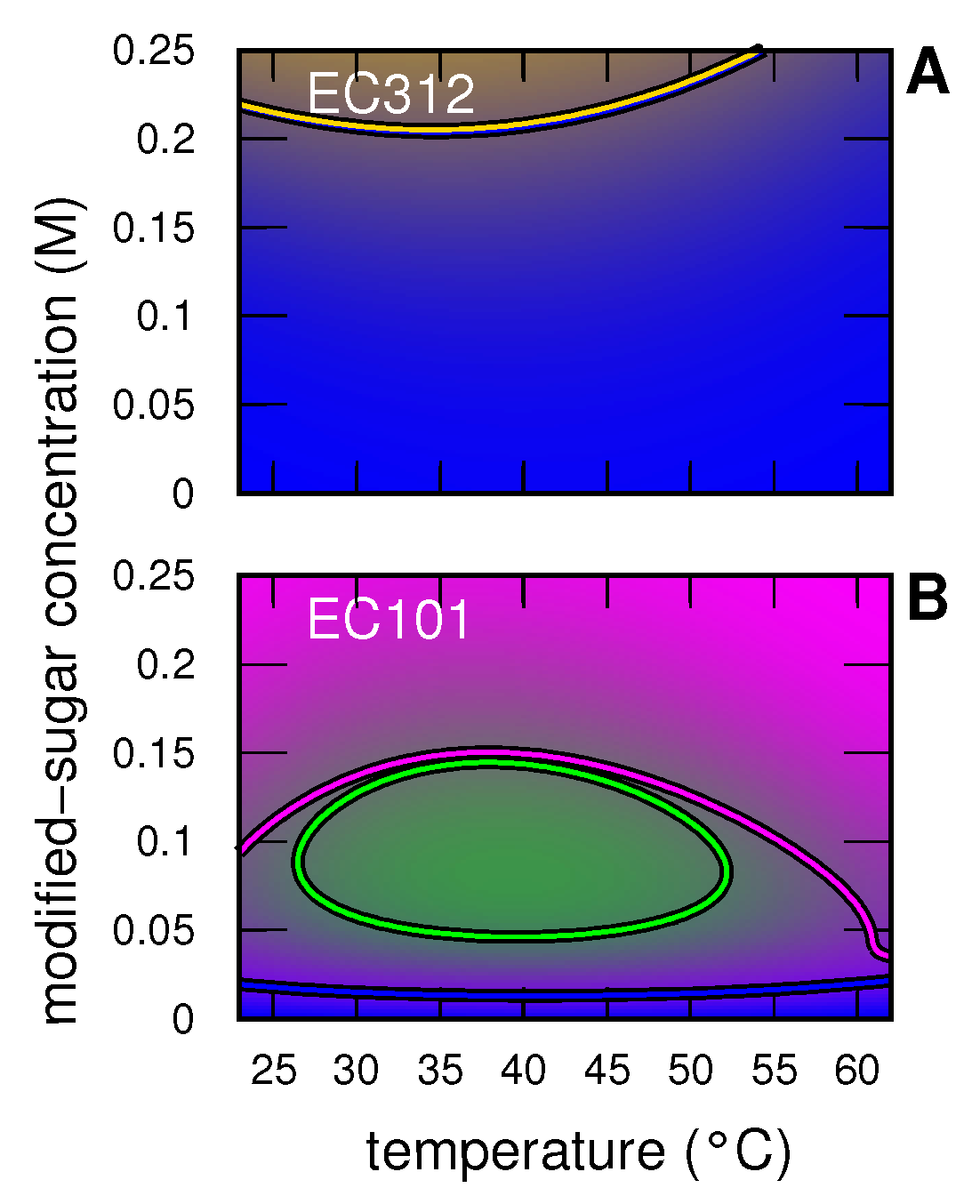
3.2. Insulin
3.2.1. Insulin without and with Modified Sugar
3.2.2. Insulin with EC312
3.2.3. Insulin with EC101
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, J.; Laurieri, N.; Delgoda, R. Proteins. In Pharmacognosy; Academic Press: Cambridge, MA, USA, 2017; pp. 477–494. [Google Scholar]
- Sane, R.; Sinz, M. Introduction of Drug Metabolism and Overview of Disease Effect on Drug Metabolism. In Drug Metabolism in Diseases; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–19. [Google Scholar]
- Mensink, M.A.; Frijlink, H.W.; van der Voort Maarschalk, K.; Hinrichs, W.L. How sugars protect proteins in the solid state and during drying (review): Mechanisms of stabilization in relation to stress conditions. Eur. J. Pharm. Biopharm. 2017, 114, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Kamerzell, T.J.; Esfandiary, R.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Protein–excipient interactions: Mechanisms and biophysical characterization applied to protein formulation development. Adv. Drug Deliv. Rev. 2011, 63, 1118–1159. [Google Scholar] [CrossRef] [PubMed]
- Ambrogi, V.; Carfagna, C.; Cerruti, P.; Marturano, V. 4-Additives in Polymers. In Modification of Polymer Properties; William Andrew Publishing: Norwich, NY, USA, 2017; pp. 87–108. [Google Scholar]
- Kontogiorgos, V. Additives in Dairy Foods: Stabilizers. In Reference Module in Food Science; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Ragoonanan, V.; Aksan, A. Protein stabilization. Transfus. Med. Hemother. 2007, 34, 246–252. [Google Scholar] [CrossRef]
- Ajito, S.; Hirai, M.; Iwase, H.; Shimizu, N.; Igarashi, N.; Ohta, N. Protective action of trehalose and glucose on protein hydration shell clarified by using X-ray and neutron scattering. Phys. B Condens. Matter 2018, 551, 249–255. [Google Scholar] [CrossRef]
- Olsson, C.; Swenson, J. The role of disaccharides for protein–protein interactions—A SANS study. Mol. Phys. 2019, 117, 3408–3416. [Google Scholar] [CrossRef] [Green Version]
- Corezzi, S.; Bracco, B.; Sassi, P.; Paolantoni, M.; Comez, L. Protein Hydration in a Bioprotecting Mixture. Life 2021, 11, 995. [Google Scholar] [CrossRef] [PubMed]
- Maycock, C.D.; Centeno, M.R.M.B.V.; Lourenço, E.C.; Dos Santos, M.H.D.; Miguel, A.S.D.C. Hexose Derivatives, Preparation and Uses Thereof. U.S. Patent US20180170955A1, 21 June 2018. [Google Scholar]
- Kumar, V.; Chari, R.; Sharma, V.K.; Kalonia, D.S. Modulation of the thermodynamic stability of proteins by polyols: Significance of polyol hydrophobicity and impact on the chemical potential of water. Int. J. Pharm. 2011, 413, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Amenitsch, H.; Rappolt, M.; Kriechbaum, M.; Mio, H.; Laggner, P.; Bernstorff, S. First performance assessment of the small-angle X-ray scattering beamline at ELETTRA. J. Sync. Rad. 1998, 5, 506–508. [Google Scholar] [CrossRef]
- Haider, R.; Sartori, B.; Radeticchio, A.; Wolf, M.; Dal Zilio, S.; Marmiroli, B.; Amenitsch, H. μDrop: A system for high-throughput small-angle X-ray scattering measurements of microlitre samples. J. Appl. Crystallogr. 2021, 54, 132–141. [Google Scholar] [CrossRef]
- Hammersley, A.P. FIT2D: A Multi-Purp. Data Reduction, Anal. Vis. Program. J. Appl. Cryst. 2016, 49, 646–652. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Ortore, M.G.; Spinozzi, F.; Mariani, P.; Paciaroni, A.; Barbosa, L.R.S.; Amenitsch, H.; Steinhart, M.; Ollivier, J.; Russo, D. Combining structure and dynamics: Non-denaturing high-pressure effect on lysozyme in solution. J. R. Soc. Interface 2009, 6, S619–S634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schellman, J.A. A simple model for solvation in mixed solvents. Applications to the stabilization and destabilization of macromolecular structures. Biohpys. Chem. 1990, 37, 121–140. [Google Scholar]
- Schellman, J.A. The thermodynamics of the solvent exchange. Biopolymers 1994, 34, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Schellman, J.A. Protein Stability in Mixed Solvents: A Balance of Contact Interaction and Excluded Volume. Biophys. J. 2003, 85, 108–125. [Google Scholar] [CrossRef] [Green Version]
- Schellman, J.A. Destabilization and stabilization of proteins. Q. Rev. Biophys. 2005, 38, 351–361. [Google Scholar] [CrossRef]
- Spinozzi, F.; Gazzillo, D.; Giacometti, A.; Mariani, P.; Carsughi, F. Interaction of proteins in solution from small angle scattering: A perturbative approach. Biophys. J. 2002, 82, 2165–2175. [Google Scholar] [CrossRef] [Green Version]
- Baldini, G.; Beretta, S.; Chirico, G.; Franz, H.; Maccioni, E.; Mariani, P.; Spinozzi, F. Salt induced association of β-lactoglobulin studied by salt light, X-ray scattering. Macromolecules 1999, 32, 6128–6138. [Google Scholar] [CrossRef]
- Moretti, P.; Mariani, P.; Ortore, M.G.; Plotegher, N.; Bubacco, L.; Beltramini, M.; Spinozzi, F. Comprehensive Structural and Thermodynamic Analysis of Prefibrillar WT α-Synuclein and Its G51D, E46K, and A53T Mutants by a Combination of Small-Angle X-ray Scattering and Variational Bayesian Weighting. J. Chem. Inf. Model. 2020, 60, 5265–5281. [Google Scholar] [CrossRef] [PubMed]
- Kell, G.S. Density, thermal expansivity, and compressibility of liquid water from 0.deg. to 150.deg. Correlations and tables for atmospheric pressure and saturation reviewed and expressed on 1968 temperature scale. J. Chem. Eng. Data 1975, 20, 97–105. [Google Scholar] [CrossRef]
- Spinozzi, F.; Ortore, M.G.; Nava, G.; Bomboi, F.; Carducci, F.; Amenitsch, H.; Bellini, T.; Sciortino, F.; Mariani, P. Gelling without Structuring: A SAXS Study of the Interactions among DNA Nanostars. Langmuir 2020, 36, 10387–10396. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.; Richard, S.; Koch, M.H.J.; Sayers, Z.; Kuprin, S.; Zaccai, G. Protein hydration in solution: Experimental observation by X-ray, neutron scattering. Proc. Natl. Acad. Sci. USA 1998, 95, 2267–2272. [Google Scholar] [CrossRef] [Green Version]
- Sinibaldi, R.; Ortore, M.G.; Spinozzi, F.; Carsughi, F.; Frielinghaus, H.; Cinelli, S.; Onori, G.; Mariani, P. Preferential hydration of lysozyme in water/glycerol mixtures: A small-angle neutron scattering study. J. Chem. Phys. 2007, 126, 235101–235109. [Google Scholar] [CrossRef] [Green Version]
- Spinozzi, F.; Mariani, P.; Ortore, M.G. Proteins in binary solvents. Biophys. Rev. 2016, 8, 87–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortore, M.; Sinibaldi, R.; Spinozzi, F.; Carbini, A.; Carsughi, F.; Mariani, P. Looking for the best experimental conditions to detail the protein solvation shell in a binary aqueous solvent via Small Angle Scattering. J. Phys. Conf. Ser. 2009, 177, 012007. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, C.L.P.; Behrens, M.A.; Pedersen, J.S.; Erlacher, K.; Otzen, D.; Pedersen, J.S. A SAXS Study of Glucagon Fibrillation. J. Mol. Biol. 2009, 387, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.P.; McDonald, I.R. Theory of Simple Liquids; Academic Press: London, UK, 1976. [Google Scholar]
- Wertheim, M.S. Exact Solution of the Percus-Yevick Integral Equation for Hard Spheres. Phys. Rev. Lett. 1963, 10, 321–323. [Google Scholar] [CrossRef]
- Ortore, M.G.; Mariani, P.; Carsughi, F.; Cinelli, S.; Onori, G.; Teixeira, J.; Spinozzi, F. Preferential solvation of lysozyme in water/ethanol mixtures. J. Chem. Phys. 2011, 135, 245103–245111. [Google Scholar] [CrossRef]
- Spinozzi, F.; Ferrero, C.; Ortore, M.G.; Antolinos, A.D.M.; Mariani, P. GENFIT: Software for the analysis of small-angle X-ray and neutron scattering data of macromolecules in-solution. J. App. Cryst. 2014, 47, 1132–1139. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.W.; Wang, J. Structure and function of heme proteins in non-native states: A mini-review. J. Inorg. Biochem. 2013, 129, 162–171. [Google Scholar] [CrossRef]
- van den Oord, A.H.A.; Wesdorp, J.J.; van Dam, A.F.; Verheij, J.A. Occurrence and Nature of Equine and Bovine Myoglobin Dimers. Eur. J. Biochem. 1969, 10, 140–145. [Google Scholar] [CrossRef]
- Nagao, S.; Osuka, H.; Yamada, T.; Uni, T.; Shomura, Y.; Imai, K.; Higuchi, Y.; Hirota, S. Structural and oxygen binding properties of dimeric horse myoglobin. Dalton Trans. 2012, 41, 11378–11385. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Ito, M.; Hirota, S.; Takada, S. Dimer domain swapping versus monomer folding in apo-myoglobin studied by molecular simulations. Phys. Chem. Chem. Phys. 2015, 17, 5006–5013. [Google Scholar] [CrossRef]
- Eliezer, D.; Chiba, K.; Tsuruta, H.; Doniach, S.; Hodgson, K.O.; Kihara, H. Evidence of an associative intermediate on the myoglobin refolding pathway. Biophys. J. 1993, 65, 912–917. [Google Scholar] [CrossRef] [Green Version]
- Baden, N.; Terazima, M. Intermolecular Interaction of Myoglobin with Water Molecules along the pH Denaturation Curve. J. Phys. Chem. B 2006, 110, 15548–15555. [Google Scholar] [CrossRef]
- Spinozzi, F.; Carsughi, F.; Mariani, P.; Saturni, L.; Bernstorff, S.; Cinelli, S.; Onori, G. Met-myoglobin association in dilute solution during pressure-induced denaturation: An analysis at pH 4.5 by high-pressure small-angle X-ray scattering. J. Phys. Chem. B 2007, 111, 3822–3830. [Google Scholar] [CrossRef] [PubMed]
- Defaye, A.B.; Ledward, D.A. Pressure-Induced Dimerization of Metmyoglobin. J. Food Sci. 1995, 60, 262–264. [Google Scholar] [CrossRef]
- Maurus, R.; Overall, C.M.; Bogumil, R.; Luo, Y.; Mauk, A.; Smith, M.; Brayer, G.D. A myoglobin variant with a polar substitution in a conserved hydrophobic cluster in the heme binding pocket. BBA-Protein Struct. Mol. Enzymol. 1997, 1341, 1–13. [Google Scholar] [CrossRef]
- Moretti, P. Innovative Methods to Investigate Intrinsically Disordered Proteins by X-ray and Neutron Scattering Techniques. Ph.D. Thesis, Life and Environmental Sciences, Ancona, Italy, 2019. [Google Scholar]
- Jeffrey, P.D.; Coates, J.H. An Equilibrium Ultracentrifuge Study of the Self-Association of Bovine Insulin. Biochemistry 1966, 5, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.; Carpenter, F.H. Zinc binding, circular dichroism, and equilibrium sedimentation studies on insulin (bovine) and several of its derivatives. Biochemistry 1974, 13, 4566–4574. [Google Scholar] [CrossRef]
- Ganim, Z.; Jones, K.C.; Tokmakoff, A. Insulin dimer dissociation and unfolding revealed by amide I two-dimensional infrared spectroscopy. Phys. Chem. Chem. Phys. 2010, 12, 3579–3588. [Google Scholar] [CrossRef]
- Uversky, V.N.; Garriques, L.N.; Millett, I.S.; Frokjaer, S.; Brange, J.; Doniach, S.; Fink, A.L. Prediction of the association state of insulin using spectral parameters. J. Pharm. Sci. 2003, 92, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Falk, B.T.; Liang, Y.; McCoy, M.A. Profiling Insulin Oligomeric States by 1H NMR Spectroscopy for Formulation Development of Ultra-Rapid-Acting Insulin. J. Pharm. Sci. 2020, 109, 922–926. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Qi, W.; Lewis, E.N.; Carpenter, J.F. Characterization of Sizes of Aggregates of Insulin Analogs and the Conformations of the Constituent Protein Molecules: A Concomitant Dynamic Light Scattering and Raman Spectroscopy Study. J. Pharm. Sci. 2016, 105, 551–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorci, M.; Belfort, G. Insulin Oligomers: Detection, Characterization and Quantification Using Different Analytical Methods. In Bio-Nanoimaging; Uversky, V.N., Lyubchenko, Y.L., Eds.; Academic Press: Boston, MA, USA, 2014; pp. 233–245. [Google Scholar]
- Nettleton, E.J.; Tito, P.; Sunde, M.; Bouchard, M.; Dobson, C.M.; Robinson, C.V. Characterization of the Oligomeric States of Insulin in Self-Assembly and Amyloid Fibril Formation by Mass Spectrometry. Biophys. J. 2000, 79, 1053–1065. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, L.S.; Nygaard, J.; Malik, L.; Hoeg-Jensen, T.; Høiberg-Nielsen, R.; Arleth, L. Structural Insight into the Self-Assembly of a Pharmaceutically Optimized Insulin Analogue Obtained by Small-Angle X-ray Scattering. Mol. Pharm. 2020, 17, 2809–2820. [Google Scholar] [CrossRef]
- O’Donoghue, S.I.; Chang, X.; Abseher, R.; Nilges, M.; Led, J.J. Unraveling the symmetry ambiguity in a hexamer: Calculation of the R6 human insulin structure. J. Biomol. NMR 2000, 16, 93–108. [Google Scholar] [CrossRef]
- Robertson, A.D.; Murphy, K.P. Protein Structure and the Energetics of Protein Stability. Chem. Rev. 1997, 97, 1251–1268. [Google Scholar] [CrossRef] [PubMed]
- Zangi, R.; Berne, B.J. Temperature Dependence of Dimerization and Dewetting of Large-Scale Hydrophobes: A Molecular Dynamics Study. J. Phys. Chem. B 2008, 112, 8634–8644. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, N.V.; Sharp, K.A. Heat Capacity in Proteins. Annu. Rev. Phys. Chem. 2005, 56, 521–548. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| kJ mol | J mol K | J mol K | |
|---|---|---|---|
| ± | 564 ± 6 | 0 ± 2 | |
| 167 ± 3 | 1600 ± 500 | 8400 ± 400 | |
| j | |||
| kJ mol | J mol K | J mol K | |
| EC312 | |||
| N | ± | ± 10 | ± 3 |
| I | ± 1 | 10 ± 10 | ± 6 |
| U | ± | ± 9 | 2 ± 3 |
| EC101 | |||
| N | ± | ± 4 | 3 ± 3 |
| I | ± | 9 ± 6 | 4 ± 7 |
| U | ± 1 | ± 7 | 0 ± 8 |
| EC311 | |||
| N | ± | 2 ± 4 | 1 ± 5 |
| I | ± | 9 ± 9 | 3 ± 7 |
| U | ± 4 | ± 2 | ± 4 |
| EC202 | |||
| N | ± 3 | ± 10 | 0 ± 4 |
| I | ± | 1 ± 4 | 7 ± 6 |
| U | ± 1 | 6 ± 9 | ± 7 |
| EC212 | |||
| N | ± | 9 ± 7 | ± 8 |
| I | ± 2 | ± 8 | 3 ± 3 |
| U | ± | 3 ± 3 | ± 4 |
| kJ mol | J mol K | J mol K | |
|---|---|---|---|
| 12 | ± | 302 ± 3 | ± 700 |
| 24 | ± 1 | 60 ± 10 | ± 800 |
| 46 | ± | 396 ± 4 | ± 700 |
| j | |||
| kJ mol | J mol K | J mol K | |
| EC312 | |||
| 1 | ± | 2 ± 5 | 0 ± 4 |
| 2 | ± | ± | 3 ± 5 |
| 4 | ± 2 | 8 ± 5 | ± 6 |
| 6 | ± | ± 5 | ± 4 |
| EC101 | |||
| 1 | ± | 6 ± 7 | 8 ± 9 |
| 2 | ± | 4 ± 9 | ± |
| 4 | ± | ± 2 | ± 2 |
| 6 | ± | ± 2 | ± 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piccinini, A.; Lourenço, E.C.; Ascenso, O.S.; Ventura, M.R.; Amenitsch, H.; Moretti, P.; Mariani, P.; Ortore, M.G.; Spinozzi, F. SAXS Reveals the Stabilization Effects of Modified Sugars on Model Proteins. Life 2022, 12, 123. https://doi.org/10.3390/life12010123
Piccinini A, Lourenço EC, Ascenso OS, Ventura MR, Amenitsch H, Moretti P, Mariani P, Ortore MG, Spinozzi F. SAXS Reveals the Stabilization Effects of Modified Sugars on Model Proteins. Life. 2022; 12(1):123. https://doi.org/10.3390/life12010123
Chicago/Turabian StylePiccinini, Astra, Eva C. Lourenço, Osvaldo S. Ascenso, Maria Rita Ventura, Heinz Amenitsch, Paolo Moretti, Paolo Mariani, Maria Grazia Ortore, and Francesco Spinozzi. 2022. "SAXS Reveals the Stabilization Effects of Modified Sugars on Model Proteins" Life 12, no. 1: 123. https://doi.org/10.3390/life12010123
APA StylePiccinini, A., Lourenço, E. C., Ascenso, O. S., Ventura, M. R., Amenitsch, H., Moretti, P., Mariani, P., Ortore, M. G., & Spinozzi, F. (2022). SAXS Reveals the Stabilization Effects of Modified Sugars on Model Proteins. Life, 12(1), 123. https://doi.org/10.3390/life12010123