1. Introduction
Microorganisms are studied in outer space for numerous reasons, including biotechnology development, model organism studies, and pathogen research. As an example of biotechnology development, the BioNutrients missions on the International Space Station (ISS) aim to use engineered
Saccharomyces cerevisiae and other microbes for the on-demand production of nutrients to be consumed by astronauts on long, deep-space missions [
1,
2]; the ISS BioRock experiment studied the ability of three microorganisms (
Sphingomonas desiccabilis,
Bacillus subtilis, and
Cupriavidus metallidurans) to aid the biomining of vanadium for in situ resource utilization [
3,
4]; on the Moonshot Artemis-1 payload,
Chlamydomonas reinhardtii algae will be grown during transit around the Moon and screened for high producers of lipids and hydrogen [
5]. For model organism research, the BioSentinel Artemis-1 CubeSat will expose wild-type and mutant
S. cerevisiae to galactic cosmic radiation and assays for metabolism and growth to better understand the biological response to deep-space radiation [
6,
7]. For pathogenesis research, the EcAMSat CubeSat used a pathogenic strain of
Escherichia coli to evaluate whether microgravity affects antibiotic resistance in low Earth orbit [
8]. In each example, microorganisms are studied as a bulk population, with each sample made up of data obtained from thousands of cells concurrently. Thus, measurements represent the averaged behavior of all cells in a sample, which can mask diverse phenotypic properties of individual cells or subpopulations [
9,
10]. Furthermore, each example relies on no more than 16 bulk samples per condition or timepoint. Single-cell analysis methods, in contrast, enable high-throughput characterizations of hundreds of thousands to millions of individual cells in the population, allowing the identification of subpopulations and rare individual cells.
There are numerous tools for analyzing and manipulating single cells, each with their own applications and limitations. Two well-established single-cell tools are flow cytometry and fluorescence-activated cell sorting (FACS), which can characterize and sort single cells by cell size and/or presence of a fluorescent marker [
11]. However, these tools are unable to characterize time-dependent phenotypes such as growth rates of clonal colonies, the cell cycle, or pathogenesis. Additionally, the analysis of cell secretions with FACS is limited, requiring additional steps such as conjugating antibodies to the cell surface, where there can be cross talk between cells without additional precautionary steps [
12,
13].
Partitioning single cells into uniform-volume compartments addresses these limitations, essentially isolating each cell in its own test tube. Standard high-throughput microwell plates have 96, 384 or even 1536 wells, but these ultimately have limited throughput when compared to high-throughput microfluidic technologies such as water-in-oil droplet emulsions, which can be used to characterize over 100,000 cells per screen [
14]. However, many applications using water-in-oil droplets require microfluidically co-encapsulating multiple reagents, such as hydrogel beads for barcoding or secretion binding, increasing the number of encapsulation steps and/or reducing the percentage of droplets containing all reagents. Additionally, the lack of continuous solution exchange in water-in-oil droplets limits the replenishment of nutrients and the elimination of cellular wastes, limiting the growth potential of encapsulated cells [
15,
16]. A similar approach uses solid hydrogel microparticles (e.g., gel microdrops), but the polymer mesh can interfere with assays, especially those involving genomic DNA [
16]. In addition, gel microdrops can limit the growth of microbes in the microparticle and can be degraded prematurely by proteases and amylases released by the encapsulated microbes [
17].
A more advanced method combines droplets and microparticles into hydrogel microparticles where an open cavity in the particle captures an aqueous droplet containing a single cell (e.g., nanovials). The hydrogel provides a surface for binding and detecting secretions, similarly to a hydrogel bead in a droplet, while avoiding the co-encapsulation step required for beads [
18,
19]. However, keeping bound cells from dislodging during long assay or growth protocols and changes in the droplet solution remains challenging. Furthermore, many microbial experiments in space, including BioNutrients, BioRock, and BioSentinel, desiccate their microorganisms prior to launch to increase long-term stabilities and to control microbe metabolic activation via rehydration [
1,
4,
20]. Encapsulating or loading rehydrated cells into water-in-oil droplets or nanovials in space would be an additional hurdle in translating these single-cell analyses to space biology research.
The alternative proposed here is fully closed hollow-shell hydrogel microparticles, or PicoShells, which can be desiccated with single microbes pre-encapsulated inside. PicoShells have a thin hydrogel shell around a hollow, aqueous core, where single cells can be encapsulated. The fully closed geometry traps the cell and its progeny inside the particle core, eliminating the need to emulsify in oil. Additionally, the hydrogel shell is porous, enabling a continuous exchange of nutrients, wastes, and assay reagents with the external environment without needing to destabilize the particle’s structure. The continuous replenishment of nutrients and the elimination of cellular wastes in the particle core allow uninhibited cell growth, more faithfully replicating suspension culture growth conditions and enabling the study of time-dependent phenotypes over days rather than only fast cellular responses. Meanwhile, the diffusion of assay reagents allows solution changes for multi-step assays (i.e., single-cell PCR or ELISA) or even multiple sequential assays on the same cells [
15,
16]. The hydrogel shell also provides a surface to conjugate various desired functional groups such as capture antibodies. Finally, PicoShells are compatible with fluorescent-activated cell sorters (FACS) such that PicoShells containing single cells or colonies can be selected based on more complex phenotypes than non-encapsulated cells. Compatibility with liquid handling and flow sorting enables the automation of assay processes to reduce crew hands-on time or to enable use in uncrewed autonomous missions such as CubeSats.
Here, we develop a method to desiccate PicoShells with encapsulated single
S. cerevisiae cells that maintains particle morphology and yeast health after storage and rehydration. Our work builds on the foundation set by van Zee et al., 2022, which introduced the PicoShell technology. Our method renders hollow-shell microparticle single-cell analyses more feasible for future space biology missions that use desiccation-tolerant microbes.
S. cerevisiae is an ideal proof-of-concept microbe as it is desiccation-tolerant, a model organism, and a useful species for biotechnology applications. Furthermore,
S. cerevisiae has been used in prior spaceflight missions such as BioNutrients and BioSentinel. Meanwhile, solid hydrogel microparticles, without voids or encapsulated cells, have been desiccated and rehydrated in oil with surfactant while maintaining a morphology similar to that pre-desiccation [
21]. We apply this desiccation strategy to PicoShells with
S. cerevisiae, desiccating particles in Novec oil with Pico-Surf surfactant under vacuum. We evaluate particle integrity via particle durability, morphology, and porosity of the hydrogel shell, and we evaluate yeast health via viability and growth potential. We further show a proof-of-concept growth-based single-cell assay on rehydrated particles.
2. Materials and Methods
2.1. Cell Culture
Desiccated wild type (WT) (YBS21-A) and mutant
rad51Δ (YBS29-1)
Saccharomyces cerevisiae strains were obtained from the NASA Ames Research Center. These strains were also used on the BioSentinel mission [
20]. Both strains are diploid prototrophic derivatives of the W303 background. The mutant
rad51Δ strain is unable to effectively repair double-stranded breaks in DNA. Both WT and
rad51Δ strains were cultured in YPD medium (Fisher BioReagents, Fair Lawn, NJ, USA) with 50 µg/mL ampicillin (Sigma-Aldrich, St. Louis, MO, USA) at 30 °C and 300 RPM.
2.2. PicoShell Fabrication and Yeast Encapsulation
Hollow-shell hydrogel microparticles (PicoShells) were fabricated using a 4-inlet T-junction microfluidic device, largely following methods from a previous study [
15]. T-junction microfluidic devices were fabricated from PDMS (polydimethylsiloxane, Sylgard
TM 184, Hayward, CA, USA) using standard soft-lithography techniques [
22]. To properly form droplets, the device channels must be hydrophobic, so each device was filled with a 2% solution of trichloro(1H,1H,2H,2H-perfluorooctyl)silane (Sigma-Aldrich, St. Louis, MO, USA) in Novec
TM 7500 engineered fluid (Novec oil, 3M
TM, St. Paul, MN, USA). After 5–10 min, the silane was washed out of the device with Novec oil, followed by vacuum aspiration to dry the device channels.
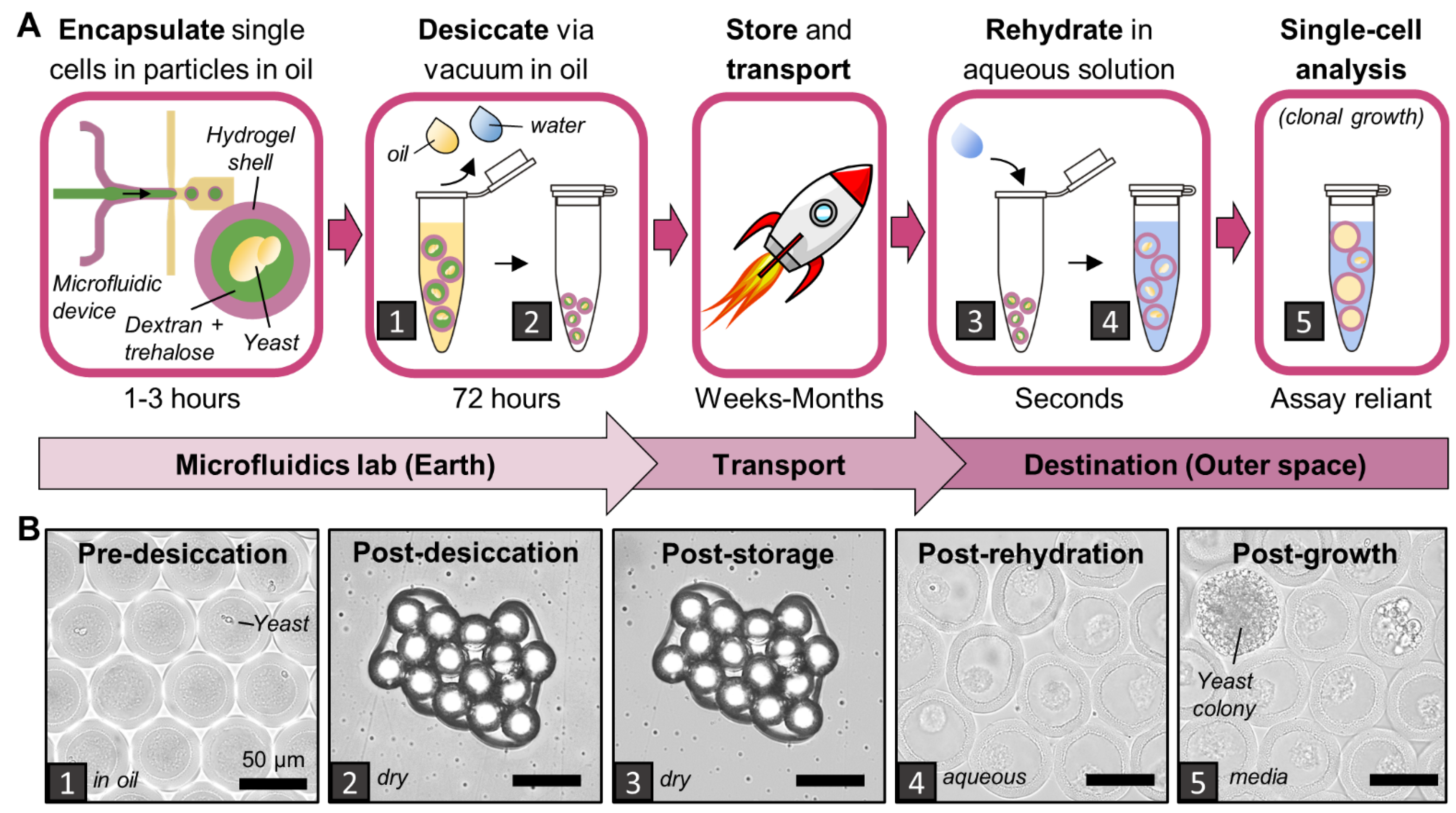
All reagents were loaded in separate syringes and pumped via syringe pumps (HA2000I, Harvard Apparatus, Holliston, MA, USA). The outer hydrogel shell is composed of 10 kDa 4-arm PEG-maleimide (10% w/w in particles, pumped at 2 μL/min, Laysan Bio, Arab, AL, USA) and DTT (1,4-dithiothreitol, 3.08 mg/mL in particles, pumped at 2 μL/min, Sigma-Aldrich, Milwaukee, WI, USA) in PBS pH 6.1. The inner liquid core is composed of 9–11 kDa dextran (20% w/w in particles, pumped at 4 μL/min, Sigma-Aldrich, Milwaukee, WI, USA) in PBS pH 6.1. The oil sheath comprises Novec oil (pumped at 35 μL/min) with 0.5% Pico-SurfTM (Sphere Fluidics, Cambridge, United Kingdom). PicoShells used in this experiment have ~70 μm outer diameter and ~11 μm shell thickness, compared with a typical S. cerevisiae radius of 2–5 μm.
S. cerevisiae were encapsulated at 8 million cells/mL in the dextran phase (average cell concentration per particle, lambda = 0.7). However, we observed that some cells were not retained in the dextran flow during particle fabrication and were found in the oil surrounding the particles, resulting in ~15% of particles containing one or more cells. Of the particles with cells, most contained single cells or 2–3 cells clumped together, which is most likely the progeny of a single cell that started budding just before or during particle fabrication. There were fewer particles containing multiple cells that were not clumped together, which is expected according to Poisson loading, but it is still potentially problematic for single-cell assays since the cells likely represent separate single cells, which could have different phenotypes. After fabrication, particles were left overnight for the hydrogel shell to fully crosslink.
WT and
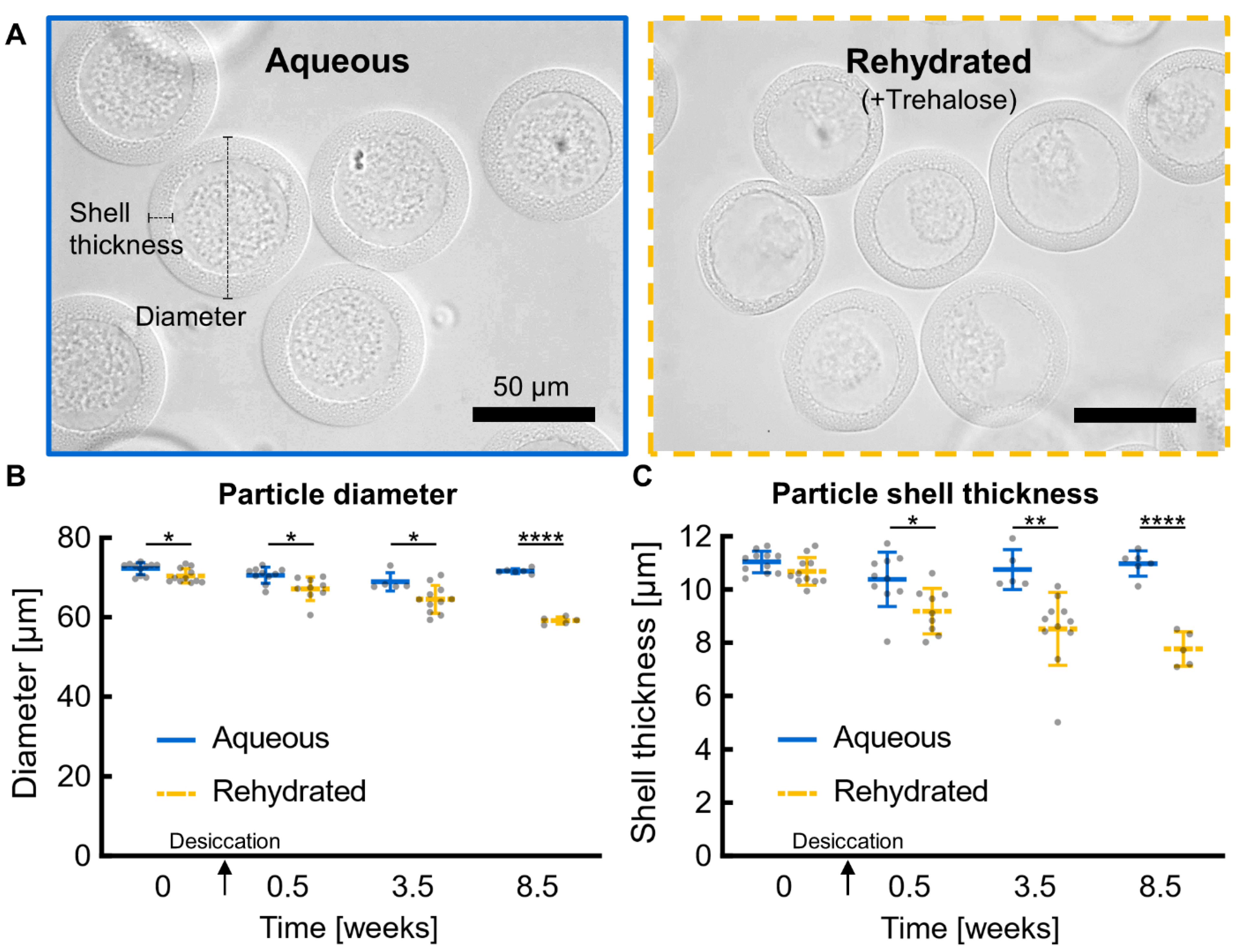
rad51Δ biological replicates for viability and growth analyses are comprised of 3 colonies picked from YPD agar plates (3 days on agar) and grown in liquid culture for 2–3 days in the cell culture conditions previously described. Yeast cells were washed 3 times with 1X PBS to remove residual medium and then resuspended in dextran with or without 10% trehalose (Fisher BioReagents, Fair Lawn, NJ, USA) for particle fabrication. Trehalose is a disaccharide and desiccation protectant commonly used to promote long-term yeast viability after desiccation [
20].
2.3. Pre-Desiccation Preparation
The day after fabrication, particles were washed with 2 mL of Novec oil over a 40 μm filter (Corning®, Durham, NC, USA) to remove non-encapsulated yeast leftover from the fabrication process. Particles retained by the filter were recovered by backwashing with 2 mL of fresh Novec oil; then, 6 μL of particles was aliquoted into polypropylene microcentrifuge tubes (Sarstedt, Nümbrecht, Germany) pre-treated with 0.1% Pluronic solution and multiple washes with deionized water. One tube of each replicate was prepared for each timepoint. Next, 10 μL of 0.5% Pico-Surf in Novec oil was added on to the particles to prevent them from beginning to dry before applying the desired desiccation method.
For the non-encapsulated yeast controls, 10 μL aliquots of yeast in a 10% trehalose solution at 107 cells/mL was plated in the bottom edge of wells in 96-well StripwellTM plates (Costar, Vernon Hills, IL, USA) following the protocol from Santa Maria et al., 2020. The non-encapsulated controls were the same 3 WT replicate yeast colonies used for encapsulation.
Separately, particles from each condition were phase transferred from Novec oil to PBS to serve as non-desiccated controls and to investigate the desiccating process in an aqueous solution. To phase transfer the particles, excess oil from fabrication was removed and Pico-BreakTM (Sphere Fluidics, Cambridge, United Kingdom) was added at a 1:1 volume ratio. PBS was added at a 3:1 volume ratio, and the mixture was vortexed and then centrifuged. Pico-Break and oil were aspirated, and the process was repeated, leaving this set of particles in PBS.
2.4. Desiccation, Storage, and Rehydration
For vacuum drying, particles in open tubes and non-encapsulated yeast in loosely covered Stripwell plates were placed in a vacuum chamber and connected to a vacuum pump (RVR003H, Dekker, Michigan City, IN, USA) for 72 h. For air drying, tubes and plates were sealed with Breathe-Easy membrane (Sigma-Aldrich, Milwaukee, WI, USA) and then exposed to ~20% relative humidity at room temperature and pressure for 3 days in a Parafilm-sealed box with Drierite desiccant (W. A. Hammond Drierite, Xenia, OH, USA). For freeze drying, samples were placed either in tubes or individual Stripwell wells inserted into tubes and frozen at −80 °C overnight. All tubes were plugged loosely with cotton balls and lyophilized for 24 h (Labconco FreeZone 4.5 L −84 °C Benchtop Freeze Dryer, Fort Scott, KS, USA).
All tubes were closed and well plates were lidded; then, desiccated particles and non-encapsulated yeast were double bagged in Ziploc bags containing Drierite desiccant and stored in a Parafilm-sealed box at room temperature (~20 °C) and ~20% relative humidity according to an Arduino-based temperature and relative humidity sensor (DHT22, Songhe, Shenzhen, China).
Cells and particles were rehydrated with 100 µL of YPD medium unless otherwise noted. Non-encapsulated yeasts were moved directly to a 30 °C incubator for 30 min. Particles were centrifuged briefly to pull the rehydrating solution through the particle clump and then vortexed and vigorously pipetted to break the clump into smaller clumps and individual particles.
2.5. Particle Morphology
Particles were imaged at key stages of desiccation: pre-desiccation, post-desiccation, post-storage, and post-rehydration. All microscopy was performed on an EVOS FL Cell Imaging Microscope (AMGTM, Mill Creek, WA, USA) except where otherwise noted. For quantitative analysis, particle diameters and shell thicknesses were measured manually in ImageJ (v1.53c) from 40× magnification images. All particle diameters and shell thicknesses were measured along a conserved axis.
2.6. Diffusion of FITC-Dextran
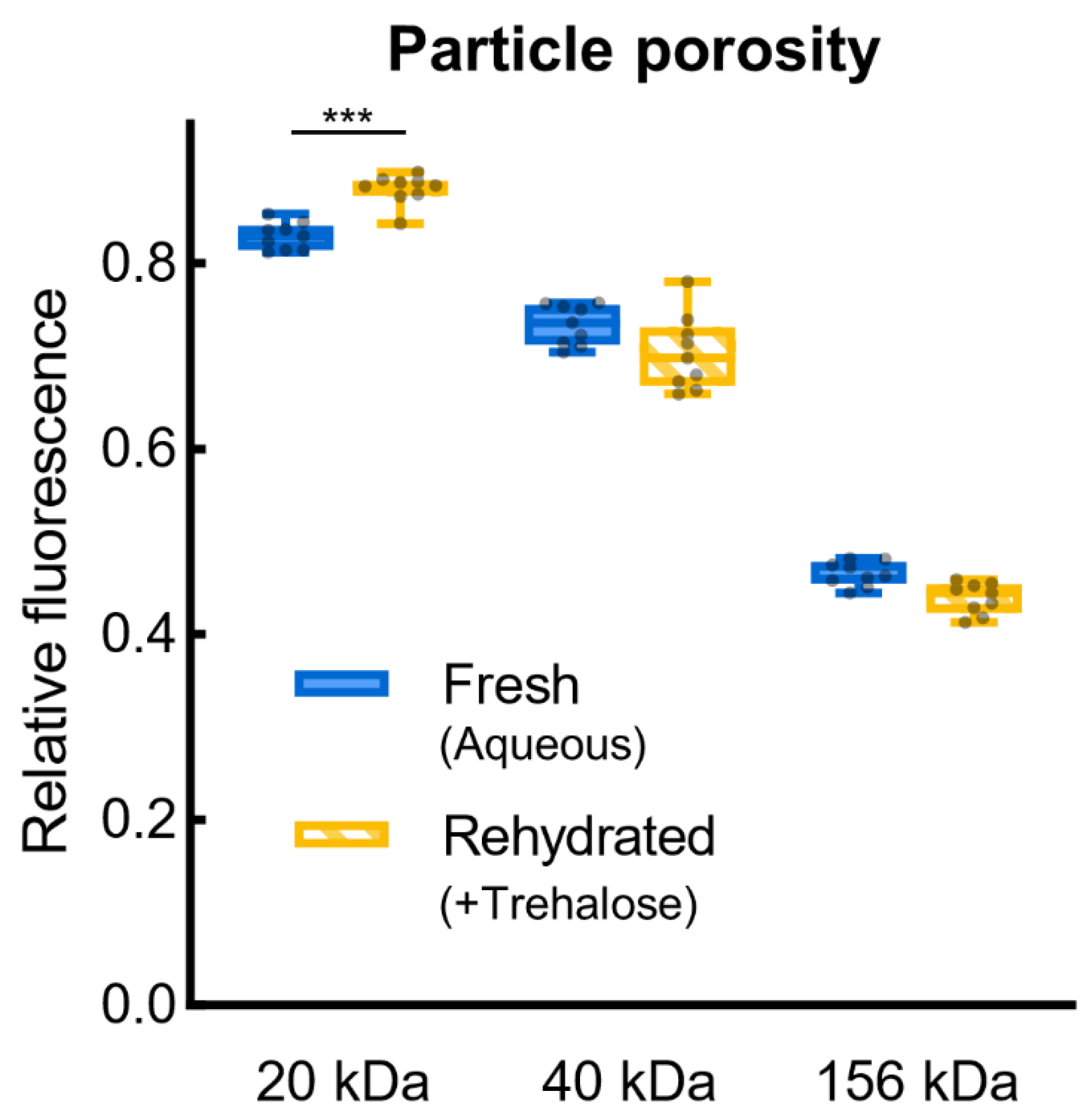
To assay diffusion across the PicoShell particles’ hydrogel shell, 20 μL of desiccated particles was rehydrated in 100 μL of PBS pH 7.4 and then mixed 1:1 with 20 kDa, 40 kDa, or 156 kDa fluorescein isothiocyanate-dextran (FITC-dextran, Sigma-Aldrich, Milwaukee, WI, USA) in deionized water to produce a 0.5 mg/mL FITC-dextran solution containing particles. This solution was immediately loaded onto a 100 μm height cellometer (Nexcelom, Lawrence, MA, USA), producing a monolayer of particles, and imaged at 20× magnification with 30 ms GFP-channel exposure at 50–80% illumination. Particles were imaged after incubation with FITC-dextran for 1–2 min (“1 min”), 20 min, 1 h, and 24 h. In ImageJ, an intensity profile cut line was measured across each particle and its immediate surroundings. The intensity within the particle was divided by the intensity outside the particle, producing a relative fluorescence inside the particle.
2.7. Viability and Growth
To assess the viability of non-encapsulated yeasts after a post-rehydration incubation (30 min), YPD medium was carefully removed and 100 μL of stain solution (PBS pH 7.4 with 10 µg/mL fluorescein diacetate (Sigma-Aldrich, Milwaukee, WI, USA) and 5 µg/mL propidium iodide (Invitrogen, Eugene, OR, USA)) was added. After incubation for 10–15 min in the dark at room temperature, an overlay image was taken at 40× magnification with brightfield (100% illumination), GFP, and RFP (60 ms exposure) channels. Live cells (stained green and unstained) and dead cells (stained red) were counted for each image using the Cell Counter plugin in ImageJ. The post-rehydration viability for each yeast culture was calculated as the number of live cells divided by the total number of cells, normalized by the viability of the same culture pre-desiccation. For example, before desiccation, a sample of one of the non-encapsulated yeast colonies had 436 live cells out of 501 total cells (87.0% viability). After desiccation and 3.5 weeks of storage, a rehydrated sample from the same yeast culture had 188 live cells out of 413 total cells (45.5% viability). The normalized viability for the 3.5-week timepoint is 45.5/87.0 = 52.3%. Normalizing helps isolate the effect of desiccation on viabilities for each condition and helps replication by accounting for natural differences in baseline viabilities.
To assess viability and growth of yeast in particles, rehydrated particles in tubes were diluted in 200 μL of YPD medium, incubated at 30 °C for 16 h, and vortexed every 6 h to ensure access to nutrients. Particles were washed 3 times with PBS, transferred to a 96-well plate (Falcon®, Durham, NC, USA), and incubated with 100 μL of stain solution for 20–40 min in the dark at room temperature. Wells were imaged on a Nikon Eclipse TI microscope using a Photometrics camera and NIS-elements AR software. The entire well was imaged with a 6.4 × 6.4 mm2 tile image at 10× magnification with brightfield, TRITC, and FITC channels; both fluorescence channels used 200 ms exposure time. The overlaid brightfield, TRITC, and FITC large-scan RGB image was manually annotated in ImageJ using the grid tool and cell counter plugin. Multiple categories of yeast in particles were annotated, including dead yeast (stained red), yeasts that are alive (stained green or unstained) but not growing robustly (fewer than 5–8 cells in the particle), particles with some yeast growth (~8–30 cells), particles full of yeast, and particles swollen with yeast (particle diameter stretched by growing yeast).
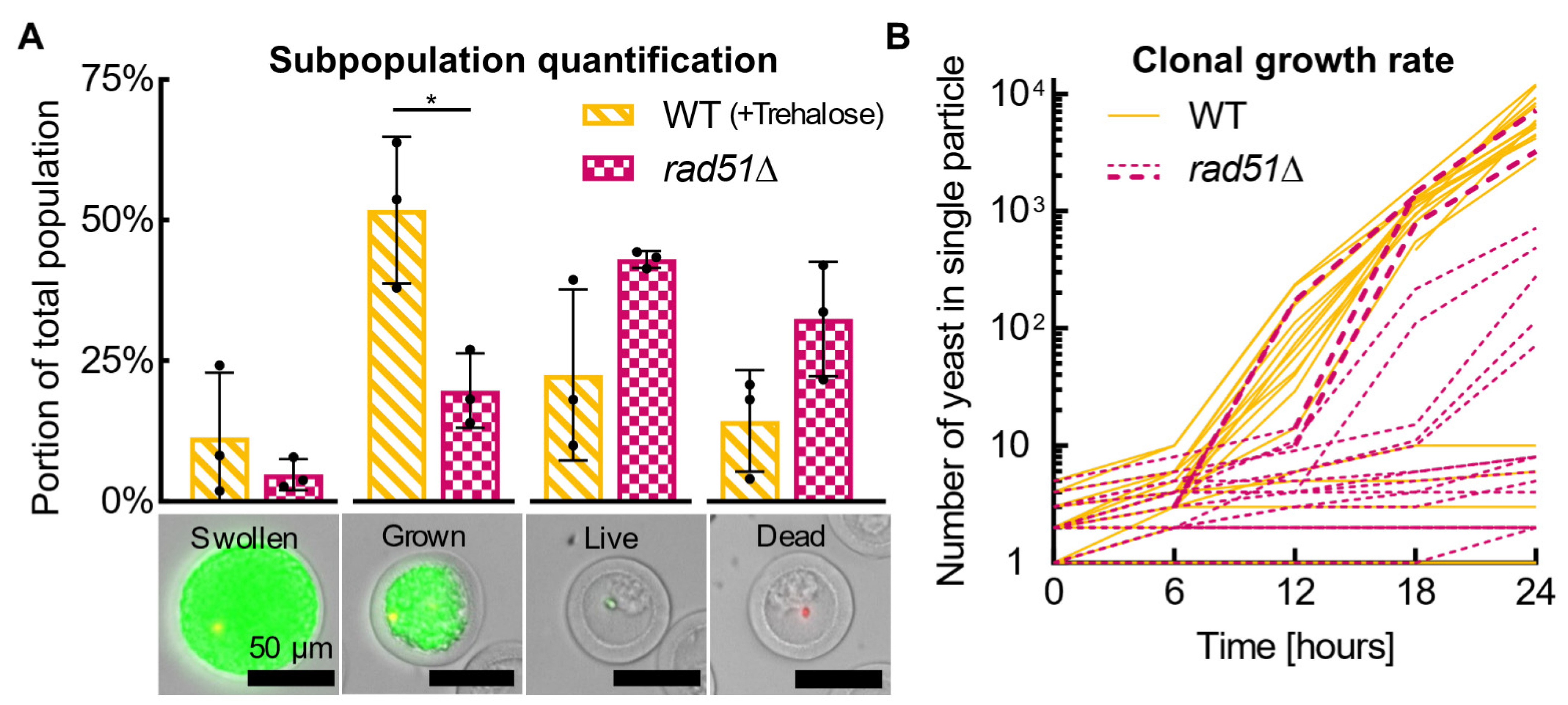
Each particle counts as one cell (i.e., a particle containing a colony of thousands of yeast cells only counts as one live cell—the parent of the colony); for viability measurements, all particles with cells are considered “live” unless they are “dead”, and the calculation and normalization is the same as for non-encapsulated cells. For yeast growth potential measurements, particles full of yeast and swollen with yeast are counted as “growing”, and normalization is applied in the same manner as viability. For subpopulation quantification, the population is divided into “swollen” (yeast stretching particle), “grown” (particle full of yeast), “live” (stained green or unstained with a single cell, a few cells, or a small clump of cells), and dead yeast. There is no normalization for subpopulation analyses.
2.8. Clonal Growth
Rehydrated particles were plated sparsely in wells with media and incubated at 30 °C without shaking. Multiple locations were selected within the well to image through time, and the well plate was handled gently to avoid shifting the particles within the well. Selected locations were imaged every 6 h for 24 h, tracking the same individual particles as the yeast within them replicated.
Each particle was tracked through the timelapse series of images, manually accounting for slight movement of particles over time. The number of yeasts in each particle was counted manually until a cell clump formed that spanned multiple focal planes. The number of yeasts in a cell clump was estimated by measuring the approximate area of the clump in ImageJ, treating that area as a circle, calculating the corresponding sphere volume, and then dividing that volume by the approximate volume of an individual yeast cell while finally multiplying it by a spherical packing factor of 0.74. The yeast’s radius was set as 2 μm, providing a yeast volume of 33.5 μm3/cell.
2.9. Statistics
All statistical tests were performed in GraphPad Prism (v8.3.0). For particle diameters, shell thickness, yeast viability, and yeast growth potential measurements, experimental conditions were compared against themselves within each timepoint with one-way ANOVAs followed by Tukey’s multiple comparisons tests. For particle shell porosity, experimental conditions were compared within and between molecular weights with a one-way ANOVA followed by Tukey’s multiple comparisons tests. Viability and growth potential measurements were also compared against themselves between 0.5 and 8.5 weeks with unpaired T-tests. Subpopulations were compared by unpaired T-tests. Throughout this paper, one asterisk (*) is p ≤ 0.05, two (**) represent p ≤ 0.01, three (***) represent p ≤ 0.001, and four (****) represent p ≤ 0.0001.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




