
Extracellular Vesicles as Surrogates for Drug Metabolism and Clearance: Promise vs. Reality


 , ,
, ,  and
and 
Abstract
:1. Introduction
1.1. Drug Metabolism and Transport
1.1.1. Drug Metabolism
1.1.2. Drug Transport
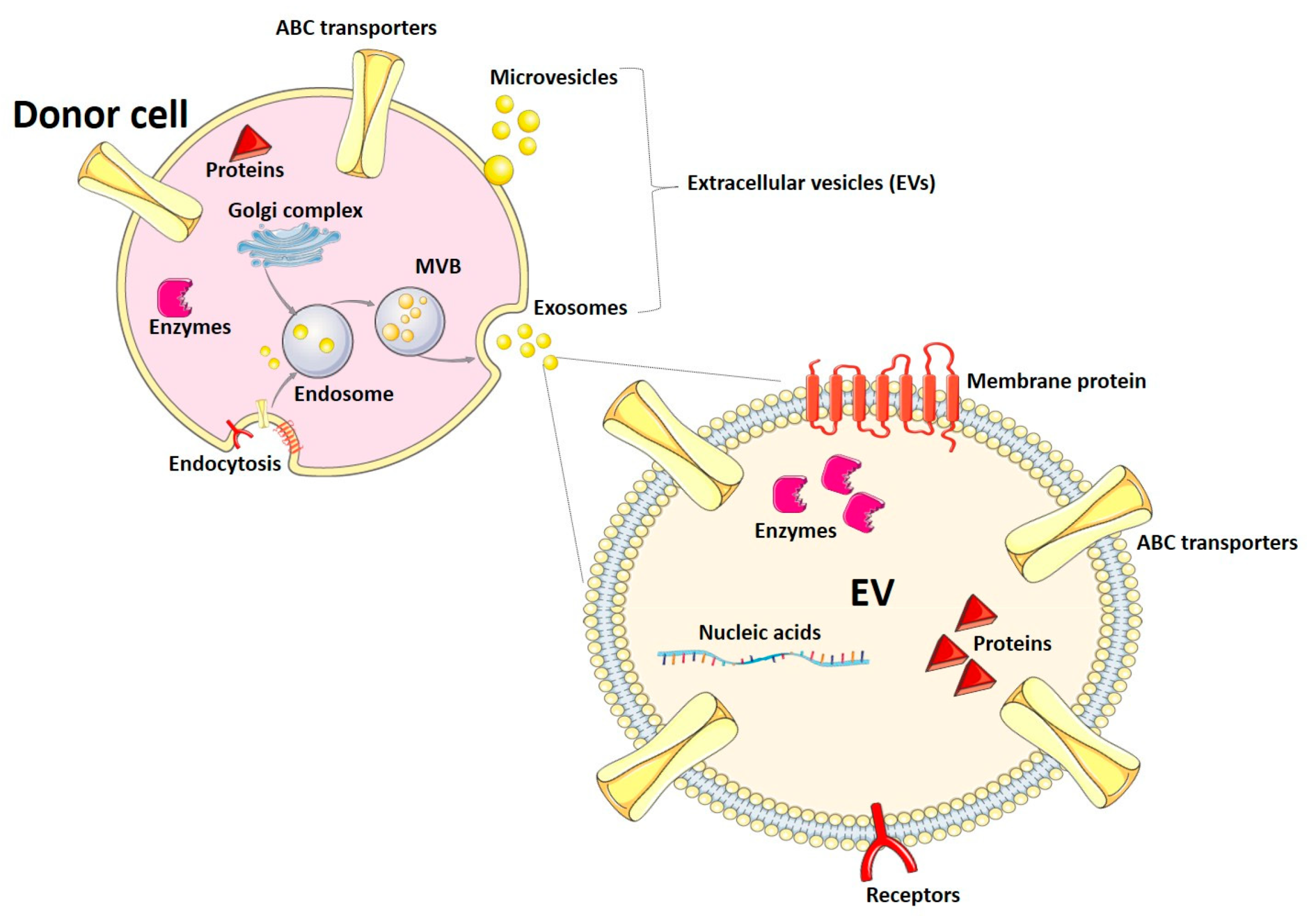
1.2. Extracellular Vesicles
2. EVs as Biomarkers of Drug Metabolism
2.1. Phase I
2.1.1. Presence of Phase I-DME in EVs
2.1.2. EVs as Dynamic Surrogates for Phase I Enzymes
2.1.3. Use of EVs for Patient Stratification
2.2. Phase II
3. EVs as Biomarkers of Drug Transport
3.1. Uptake Transporters
3.2. Efflux Transporters
3.2.1. Plasma and Serum EVs
3.2.2. Urinary EVs
4. Future Perspectives and Challenges
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rathore, S.S. Association of Serum Digoxin Concentration and Outcomes in Patients with Heart Failure. JAMA 2003, 289, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Morita, M.; Miyaji, F. Population Pharmacokinetics and Optimization of the Dosing Regimen of Digoxin in Adult Patients. J. Pharm. Health Care Sci. 2015, 1, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, M.E.; Parvez, M.M.; Shin, J.G. Clinical Implementation of Pharmacogenomics for Personalized Precision Medicine: Barriers and Solutions. J. Pharm. Sci. 2017, 106, 2368–2379. [Google Scholar] [CrossRef] [Green Version]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T. Models and mechanism of cytochrome P450. In Cytochrome P450: Structure Mechanism and Biochemistry, 3rd ed.; Ortiz de Montellano, P.R., Ed.; Springer: Boston, MA, USA, 2005. [Google Scholar] [CrossRef]
- Ding, X.; Kaminsky, L.S. Human Extrahepatic Cytochrome P450: Function in Xenobiotic Metabolism and Tissue-Selective Chemical Toxicity in the Respiratory and Gastrointestinal Tracts. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Krishna, D.R.; Klotz, U. Extrahepatic Metabolism of Drugs in Humans. Clin. Pharmacokinet. 1994, 26, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.V.; Eddershaw, P.J.; Dickins, M.; Tarbit, M.H.; Goldfarb, P.S. Structural determinants of cytochrome P450 substrate specificity, binding affinity and catalytic rate. Chem. Biol. Interact. 1998, 115, 175–199. [Google Scholar] [CrossRef]
- Okey, A.B. Enzyme induction in the cytochrome P-450 system. Pharmacol. Ther. 1990, 45, 241–298. [Google Scholar] [CrossRef]
- Barry, M.; Feely, J. Enzyme induction and inhibition. Pharmacol. Ther. 1990, 48, 71–94. [Google Scholar] [CrossRef]
- Willson, T.M.; Kliewer, S.A. Pxr, car and drug metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Wiesinger, H.; Klein, S.; Rottmann, A.; Nowotny, B.; Riecke, K.; Gashaw, I.; Brudny-Klöppel, M.; Fricke, R.; Höchel, J.; Friedrich, C. The Effects of Weak and Strong CYP3A Induction by Rifampicin on the Pharmacokinetics of Five Progestins and Ethinylestradiol Compared to Midazolam. Clin. Pharmacol. Ther. 2020, 108, 798–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, T.; Fujii-Kuriyama, Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and1B1. Cancer Sci. 2004, 95, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Goedtke, L.; Sprenger, H.; Hofmann, U.; Schmidt, F.F.; Hammer, H.S.; Zanger, U.M.; Poetz, O.; Seidel, A.; Braeuning, A.; Hessel-Pras, S. Polycyclic Aromatic Hydrocarbons Activate the Aryl Hydrocarbon Receptor and the Constitutive Androstane Receptor to Regulate Xenobiotic Metabolism in Human Liver Cells. Int. J. Mol. Sci. 2020, 22, 372. [Google Scholar] [CrossRef] [PubMed]
- Tukey, R.H.; Strassburg, C.P. Human UDP-Glucuronosyltransferases: Metabolism, Expression, and Disease. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 581–616. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Gamage, N.; Barnett, A.; Hempel, N.; Duggleby, R.G.; Windmill, K.F.; Martin, J.L.; McManus, M.E. Human Sulfotransferases and Their Role in Chemical Metabolism. Toxicol. Sci. 2005, 90, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Klimas, R.; Mikus, G. Morphine-6-glucuronide is responsible for the analgesic effect after morphine administration: A quantitative review of morphine, morphine-6-glucuronide, and morphine-3-glucuronide. Br. J. Anaesth. 2014, 113, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Köhle, C.; Bock, K.W. Coordinate regulation of human drug-metabolizing enzymes, and conjugate transporters by the Ah receptor, pregnane X receptor and constitutive androstane receptor. Biochem. Pharmacol. 2009, 77, 689–699. [Google Scholar] [CrossRef]
- Gupta, E.; Wang, X.; Ramirez, J.; Ratain, M.J. Modulation of glucuronidation of SN-38, the active metabolite of irinotecan, by valproic acid and phenobarbital. Cancer Chemother. Pharmacol. 1997, 39, 440–444. [Google Scholar] [CrossRef]
- Zhong, S.; Huang, M.; Yang, X.; Liang, L.; Wang, Y.; Romkes, M.; Duan, W.; Chan, E.; Zhou, S.-F. Relationship of glutathione S-transferase genotypes with side-effects of pulsed cyclophosphamide therapy in patients with systemic lupus erythematosus. Br. J. Clin. Pharmacol. 2006, 62, 457–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langouet, S.; Morel, F.; Meyer, D.J.; Fardel, O.; Corcos, L.; Ketterer, B.; Guillouzo, A. A comparison of the effect of inducers on the expression of glutathione-S-transferases in the liver of the intact rat and in hepatocytes in primary culture. Hepatology 1996, 23, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Goren, A.; Dhurat, R.; Agrawal, S.; Sinclair, R.; Trüeb, R.M.; Vañó-Galván, S.; Chen, G.; Tan, Y.; Kovacevic, M.; et al. Tretinoin enhances minoxidil response in androgenetic alopecia patients by upregulating follicular sulfotransferase enzymes. Dermatol. Ther. 2019, 32, e12915. [Google Scholar] [CrossRef] [PubMed]
- Kodama, S.; Hosseinpour, F.; Goldstein, J.A.; Negishi, M. Liganded pregnane X receptor represses the human sulfotransferase SULT1E1 promoter through disrupting its chromatin structure. Nucleic Acids Res. 2011, 39, 8392–8403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.L.; Strom, S.C.; Ellis, E.; Duanmu, Z.; Fu, J.; Duniec-Dmuchowski, Z.; Falany, C.N.; Falany, J.L.; Kocarek, T.A.; Runge-Morris, M. Positive and Negative Regulation of Human Hepatic Hydroxysteroid Sulfotransferase (SULT2A1) Gene Transcription by Rifampicin: Roles of Hepatocyte Nuclear Factor 4α and Pregnane X Receptor. J. Pharmacol. Exp. Ther. 2007, 323, 586–598. [Google Scholar] [CrossRef]
- Aleksunes, L.M.; Klaassen, C.D. Coordinated Regulation of Hepatic Phase I and II Drug-Metabolizing Genes and Transporters using AhR-, CAR-, PXR-, PPARα-, and Nrf2-Null Mice. Drug Metab. Dispos. 2012, 40, 1366–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzagalli, M.D.; Bensimon, A.; Superti-Furga, G. A guide to plasma membrane solute carrier proteins. FEBS J. 2020, 288, 2784–2835. [Google Scholar] [CrossRef]
- Wang, D.S.; Jonker, J.W.; Kato, Y.; Kusuhara, H.; Schinkel, A.H.; Sugiyama, Y. Involvement of Organic Cation Transporter 1 in Hepatic and Intestinal Distribution of Metformin. J. Pharmacol. Exp. Ther. 2002, 302, 510–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellick, K. Organic Ion Transporters and Statin Drug Interactions. Curr. Atheroscler. Rep. 2017, 19, 65. [Google Scholar] [CrossRef] [PubMed]
- van de Steeg, E.; van Esch, A.; Wagenaar, E.; Kenworthy, K.E.; Schinkel, A.H. Influence of Human OATP1B1, OATP1B3, and OATP1A2 on the Pharmacokinetics of Methotrexate and Paclitaxel in Humanized Transgenic Mice. Clin. Cancer Res. 2012, 19, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Iusuf, D.; van Esch, A.; Hobbs, M.; Taylor, M.; Kenworthy, K.E.; van de Steeg, E.; Wagenaar, E.; Schinkel, A.H. Murine Oatp1a/1b Uptake Transporters Control Rosuvastatin Systemic Exposure without Affecting Its Apparent Liver Exposure. Mol. Pharmacol. 2013, 83, 919–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, T.; Akiyoshi, T.; Tsuchitani, T.; Kataoka, H.; Araki, N.; Yajima, K.; Katayama, K.; Imaoka, A.; Ohtani, H. Inhibitory Effects of Cranberry Juice and Its Components on Intestinal OATP1A2 and OATP2B1: Identification of Avicularin as a Novel Inhibitor. J. Agric. Food Chem. 2022, 70, 3310–3320. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The International Transporter Consortium; Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D.; Aleksunes, L.M. Xenobiotic, Bile Acid, and Cholesterol Transporters: Function and Regulation. Pharmacol. Rev. 2010, 62, 1–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez de Marcos, J.C.; Pérez-Pineda, P.L.; Méndez-Morales, S.T.; Arellano-Mendoza, M.G.; Torres Espíndola, L.M. ABC transporter superfamily. An updated overview, relevance in cancer multidrug resistance and perspectives with personalized medicine. Mol. Biol. Rep. 2021, 48, 1883–1901. [Google Scholar] [CrossRef]
- Fenner, K.; Troutman, M.; Kempshall, S.; Cook, J.; Ware, J.; Smith, D.; Lee, C. Drug–Drug Interactions Mediated Through P-Glycoprotein: Clinical Relevance and In Vitro–In Vivo Correlation Using Digoxin as a Probe Drug. Clin. Pharmacol. Ther. 2008, 85, 173–181. [Google Scholar] [CrossRef]
- Nies, A.T.; Keppler, D. The apical conjugate efflux pump ABCC2 (MRP2). Pflugers Arch. Eur. J. Physiol. 2006, 453, 643–659. [Google Scholar] [CrossRef]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef]
- Robey, R.W.; Polgar, O.; Deeken, J.; To, K.W.; Bates, S.E. ABCG2: Determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007, 26, 39–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Z.; Bikadi, Z.; Rosenberg, M.F.; Mao, Q. Structure and Function of the Human Breast Cancer Resistance Protein (BCRP/ABCG2). Curr. Drug Metab. 2010, 11, 603–617. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- D’Souza-Schorey, C.; Schorey, J.S. Regulation and mechanisms of extracellular vesicle biogenesis and secretion. Essays Biochem. 2018, 62, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Saxena, N.; Vrana, M.; Zhang, H.; Kumar, V.; Billington, S.; Khojasteh, C.; Heyward, S.; Unadkat, J.D.; Prasad, B. Targeted LC-MS/MS Proteomics-Based Strategy to Characterize in Vitro Models Used in Drug Metabolism and Transport Studies. Anal. Chem. 2018, 90, 11873–11882. [Google Scholar] [CrossRef] [PubMed]
- Conde-Vancells, J.; Rodriguez-Suarez, E.; Embade, N.; Gil, D.; Matthiesen, R.; Valle, M.; Elortza, F.; Lu, S.C.; Mato, J.M.; Falcon-Perez, J.M. Characterization and Comprehensive Proteome Profiling of Exosomes Secreted by Hepatocytes. J. Proteome Res. 2008, 7, 5157–5166. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Sinha, N.; Gerth, K.A.; Rahman, M.A.; Yallapu, M.M.; Midde, N.M. Specific packaging and circulation of cytochromes P450, especially 2E1 isozyme, in human plasma exosomes and their implications in cellular communications. Biochem. Biophys. Res. Commun. 2017, 491, 675–680. [Google Scholar] [CrossRef]
- Rowland, A.; Ruanglertboon, W.; Dyk, M.; Wijayakumara, D.; Wood, L.S.; Meech, R.; Mackenzie, P.I.; Rodrigues, A.D.; Marshall, J.; Sorich, M.J. Plasma extracellular nanovesicle (exosome)-derived biomarkers for drug metabolism pathways: A novel approach to characterize variability in drug exposure. Br. J. Clin. Pharmacol. 2018, 85, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Suárez, E.; Gonzalez, E.; Hughes, C.; Conde-Vancells, J.; Rudella, A.; Royo, F.; Palomo, L.; Elortza, F.; Lu, S.C.; Mato, J.M.; et al. Quantitative proteomic analysis of hepatocyte-secreted extracellular vesicles reveals candidate markers for liver toxicity. J. Proteom. 2014, 103, 227–240. [Google Scholar] [CrossRef]
- Cho, Y.-E.; Mezey, E.; Hardwick, J.P.; Salem, N.; Clemens, D.L.; Song, B.-J. Increased ethanol-inducible cytochrome P450-2E1 and cytochrome P450 isoforms in exosomes of alcohol-exposed rodents and patients with alcoholism through oxidative and endoplasmic reticulum stress. Hepatol. Commun. 2017, 1, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Kodidela, S.; Sinha, N.; Haque, S.; Shukla, P.K.; Rao, R.; Kumar, S. Plasma exosomes exacerbate alcohol- and acetaminophen-induced toxicity via CYP2E1 pathway. Sci. Rep. 2019, 9, 6571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, O.; Morrison, E.E.; Tranter, J.D.; Starkey Lewis, P.; Toor, I.S.; Srivastava, A.; Sargeant, R.; Rollison, H.; Matchett, K.P.; Kendall, T.J.; et al. Transfer of hepatocellular microRNA regulates cytochrome P450 2E1 in renal tubular cells. EBioMedicine 2020, 62, 103092. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.D.; Dyk, M.; Sorich, M.J.; Fahmy, A.; Useckaite, Z.; Newman, L.A.; Kapetas, A.J.; Mounzer, R.; Wood, L.S.; Johnson, J.G.; et al. Exploring the Use of Serum-Derived Small Extracellular Vesicles as Liquid Biopsy to Study the Induction of Hepatic Cytochromes P450 and Organic Anion Transporting Polypeptides. Clin. Pharmacol. Ther. 2021, 110, 248–258. [Google Scholar] [CrossRef]
- Rodrigues, A.D.; Wood, L.S.; Vourvahis, M.; Rowland, A. Leveraging Human Plasma-Derived Small Extracellular Vesicles as Liquid Biopsy to Study the Induction of Cytochrome P450 3A4 by Modafinil. Clin. Pharmacol. Ther. 2021, 111, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Achour, B.; Al-Majdoub, Z.M.; Grybos-Gajniak, A.; Lea, K.; Kilford, P.; Zhang, M.; Knight, D.; Barber, J.; Schageman, J.; Rostami-Hodjegan, A. Liquid Biopsy Enables Quantification of the Abundance and Interindividual Variability of Hepatic Enzymes and Transporters. Clin. Pharmacol. Ther. 2020, 109, 222–232. [Google Scholar] [CrossRef]
- Achour, B.; Gosselin, P.; Terrier, J.; Gloor, Y.; Al-Majdoub, Z.M.; Polasek, T.M.; Daali, Y.; Rostami-Hodjegan, A.; Reny, J. Liquid Biopsy for Patient Characterization in Cardiovascular Disease: Verification against Markers of Cytochrome P450 and P-Glycoprotein Activities. Clin. Pharmacol. Ther. 2022, 111, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Pridgeon, C.S.; Johansson, I.; Ingelman-Sundberg, M. Liquid biopsies or therapeutic drug monitoring for CYP activity profile determination. Clin. Pharmacol. Ther. 2022, 112, 1000–1003. [Google Scholar] [CrossRef]
- Raj, D.A.A.; Fiume, I.; Capasso, G.; Pocsfalvi, G. A multiplex quantitative proteomics strategy for protein biomarker studies in urinary exosomes. Kidney Int. 2012, 81, 1263–1272. [Google Scholar] [CrossRef] [Green Version]
- Pisitkun, T.; Shen, R.F.; Knepper, M.A. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA 2004, 101, 13368–13373. [Google Scholar] [CrossRef]
- Gonzales, P.A.; Pisitkun, T.; Hoffert, J.D.; Tchapyjnikov, D.; Star, R.A.; Kleta, R.; Wang, N.S.; Knepper, M.A. Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes. J. Am. Soc. Nephrol. 2008, 20, 363–379. [Google Scholar] [CrossRef] [Green Version]
- Margaillan, G.; Rouleau, M.; Fallon, J.K.; Caron, P.; Villeneuve, L.; Turcotte, V.; Smith, P.C.; Joy, M.S.; Guillemette, C. Quantitative Profiling of Human Renal UDP-glucuronosyltransferases and Glucuronidation Activity: A Comparison of Normal and Tumoral Kidney Tissues. Drug Metab. Dispos. 2015, 43, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, A.D.; Lai, Y.; Shen, H.; Varma, M.V.S.; Rowland, A.; Oswald, S. Induction of Human Intestinal and Hepatic Organic Anion Transporting Polypeptides: Where Is the Evidence for Its Relevance in Drug-Drug Interactions? Drug Metab. Dispos. 2019, 48, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Campagno, R.V.; Nosetto, E.C.; Brandoni, A.; Torres, A.M. Hepatic and renal expression of Oatp1 in obstructive uropathy. First detection of Oatp1 in urine, a potential biomarker. Clin. Exp. Pharmacol. Physiol. 2021, 48, 987–995. [Google Scholar] [CrossRef]
- Blijdorp, C.J.; Tutakhel, O.A.Z.; Hartjes, T.A.; van den Bosch, T.P.P.; van Heugten, M.H.; Rigalli, J.P.; Willemsen, R.; Musterd-Bhaggoe, U.M.; Barros, E.R.; Carles-Fontana, R.; et al. Comparing Approaches to Normalize, Quantify, and Characterize Urinary Extracellular Vesicles. J. Am. Soc. Nephrol. 2021, 32, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Kunin, M.; Holtzman, E.J.; Melnikov, S.; Dinour, D. Urinary organic anion transporter protein profiles in AKI. Nephrol. Dial. Transplant. 2011, 27, 1387–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotanda, K.; Hirota, T.; Saito, J.; Fukae, M.; Egashira, Y.; Izumi, N.; Deguchi, M.; Kimura, M.; Matsuki, S.; Irie, S.; et al. Circulating intestine-derived exosomal miR-328 in plasma, a possible biomarker for estimating BCRP function in the human intestines. Sci. Rep. 2016, 6, 32299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Pan, Y.-Z.; Seigel, G.M.; Hu, Z.-H.; Huang, M.; Yu, A.-M. Breast cancer resistance protein BCRP/ABCG2 regulatory microRNAs (hsa-miR-328, -519c and -520h) and their differential expression in stem-like ABCG2+ cancer cells. Biochem. Pharmacol. 2011, 81, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Mizutani, K.; Kameyama, K.; Kawakami, K.; Fujita, Y.; Nakane, K.; Kanimoto, Y.; Ehara, H.; Ito, H.; Seishima, M.; et al. Serum exosomal P-glycoprotein is a potential marker to diagnose docetaxel resistance and select a taxoid for patients with prostate cancer. Urol. Oncol. 2015, 33, 385.e15–385.e20. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Enzyme | Detection Level | Sample | Regulation | References |
|---|---|---|---|---|
| CYP1A1 | Protein Protein Protein | Serum Plasma (rat) Plasma (mice) | Ethanol ↑ Ethanol ↑ Ethanol ↑ | [52] |
| CYP1A2 | Protein Protein Protein mRNA, protein | Serum Plasma (rat) Plasma (mice) Plasma | Ethanol ↑ Ethanol ↑ Ethanol ↑ ─ | [52] [50,57] |
| CYP1B1 | mRNA, protein | Plasma | ─ | [49] |
| CYP2A1 | Protein Protein | Culture medium (rat hepatocytes) Culture medium (rat hepatocytes) | galN ↑ ─ | [51] [48] |
| CYP2A2 | Protein | Plasma (rat) | Ethanol ↑ | [52] |
| CYP2A3 | Protein | Plasma (rat) | Ethanol ↑ | [52] |
| CYP2A5 | Protein | Plasma (mice) | Ethanol ↑ | [52] |
| CYP2A6 | Protein mRNA, protein | Serum Plasma | Ethanol ↑ ─ | [52] [49,57] |
| CYP2B2 | Protein | Culture medium (rat hepatocytes) | galN ↑ | [51] |
| CYP2B3 | Protein Protein | Culture medium (rat hepatocytes) Culture medium (rat hepatocytes) | galN ↑ ─ | [51] [48] |
| CYP2B6 | Protein | Plasma | ─ | [50] |
| CYP2C8 | mRNA, protein | Plasma | ─ | [50] |
| CYP2C9 | mRNA, protein | Plasma | ─ | [50,57] |
| CYP2C11 | Protein Protein | Culture medium (rat hepatocytes) Culture medium (rat hepatocytes) | galN ↑ ─ | [51] [48] |
| CYP2C19 | mRNA, protein | Plasma | ─ | [50,57] |
| CYP2C23 | Protein | Culture medium (rat hepatocytes) | galN ↑ | [51] |
| CYP2D1 | Protein | Culture medium (rat hepatocytes) | ─ | [48] |
| CYP2D3 | Protein | Culture medium (rat hepatocytes) | ─ | [48] |
| CYP2D4 | Protein | Culture medium (rat hepatocytes) | LPS ↓ | [51] |
| CYP2D6 | mRNA, protein | Plasma | ─ | [50,55,57] |
| CYP2D10 | Protein Protein | Culture medium (rat hepatocytes) Culture medium (rat hepatocytes) | galN ↑ ─ | [51] [48] |
| CYP2D18 | Protein Protein | Culture medium (rat hepatocytes) Culture medium (rat hepatocytes) | galN ↑ ─ | [51] [48] |
| CYP2D26 | Protein | Culture medium (rat hepatocytes) | ─ | [48] |
| CYP2E1 | Protein Protein Protein mRNA, protein | Serum Plasma (rat) Plasma (mice) Plasma | Ethanol ↑ Ethanol ↑ Ethanol ↑ ─ | [52] [49,50,57] |
| CYP2J2 | Protein | Plasma | ─ | [50] |
| CYP3A4 | mRNA, protein, enzymatic activity mRNA, protein | Plasma Plasma | Rifampicin ↑ ─ | [50,55,57] [49] |
| CYP3A5 | Protein mRNA, protein | Culture medium (human hepatocytes) Plasma | ─ ─ | [47] [50,57] |
| CYP4A | Protein Protein Protein | Serum Plasma (rat) Plasma (mice) | ─ ─ ─ | [52] |
| CYP4B | Protein Protein Protein | Serum Plasma (rat) Plasma (mice) | Ethanol↑ Ethanol↑ Ethanol↑ | [52] |
| Enzyme | Detection Level | Sample | Regulation | References |
|---|---|---|---|---|
| GSTA1 | Protein mRNA Protein | Culture medium (rat hepatocytes) Plasma Urine | LPS ↑ ─ ─ | [51] [57] [60,61,62] |
| GSTA2 | mRNA Protein | Plasma Urine | ─ ─ | [57] [60,61] |
| GSTA3 | Protein | Urine | ─ | [61] |
| GSTM1 | Protein Protein mRNA | Culture medium (rat hepatocytes) Culture medium (rat hepatocytes) Plasma | galN ↑ ─ ─ | [51] [48] [57] |
| GSTM2 | Protein mRNA Protein | Culture medium (rat hepatocytes) Plasma Urine | galN ↓ LPS ↓ ─ ─ | [51] [57] [60,61] |
| GSTM3 | Protein | Urine | ─ | [60,61] |
| GSTM4 | Protein mRNA | Culture medium (rat hepatocytes) Plasma | LPS ↑ ─ | [51] [57] |
| GSTM5 | mRNA Protein | Plasma Urine | ─ ─ | [57] [60] |
| GSTP1 | Protein | Urine | ─ | [60,62] |
| GSTT1 | mRNA | Plasma | ─ | [57] |
| SULT1A1 | Protein mRNA | Culture medium (rat hepatocytes) Plasma | ─ ─ | [48] [57] |
| SULT1A2 | mRNA | Plasma | ─ | [57] |
| SULT1A4 | mRNA | Plasma | ─ | [57] |
| SULT1B1 | mRNA | Plasma | ─ | [57] |
| SULT1C2 | mRNA | Plasma | ─ | [57] |
| SULT1E1 | Protein mRNA | Culture medium (rat hepatocytes) Plasma | galN ↑ ─ | [51] [57] |
| SULT1E2 | Protein | Culture medium (rat hepatocytes) | galN ↑ LPS ↑ | [51] |
| SULT1E3 | Protein | Culture medium (rat hepatocytes) | galN ↑ | [51] |
| SULT2A1 | Protein Protein mRNA | Culture medium (human hepatocytes) Culture medium (rat hepatocytes) Plasma | ─ galN ↓ LPS ↓ ─ | [47] [51] [57] |
| UGT1A1 | Protein mRNA, protein | Culture medium (rat hepatocytes) Plasma | galN ↑ ─ | [51] [50,57] |
| UGT1A2 | Protein Protein | Culture medium (rat hepatocytes) Plasma | galN ↑ ─ | [51] [50] |
| UGT1A3 | Protein mRNA, protein | Culture medium (rat hepatocytes) Plasma | galN ↑ ─ | [51] [50,57] |
| UGT1A4 | mRNA, protein | Plasma | ─ | [50,57] |
| UGT1A6 | Protein Protein Protein | Culture medium (rat hepatocytes) Plasma Culture medium (rat hepatocytes) | galN ↑ ─ ─ | [51] [50] [48] |
| UGT1A7 | Protein | Culture medium (rat hepatocytes) | galN ↑ | [51] |
| UGT1A8 | Protein mRNA Protein | Culture medium (rat hepatocytes) Plasma Culture medium (rat hepatocytes) | galN ↑ ─ ─ | [51] [57] [48] |
| UGT1A9 | mRNA, protein | Plasma | ─ | [50,57] |
| UGT1A10 | mRNA | Plasma | ─ | [57] |
| UGT2A3 | mRNA | Plasma | ─ | [57] |
| UGT2B1 | Protein | Culture medium (rat hepatocytes) | ─ | [48] |
| UGT2B2 | Protein Protein | Culture medium (rat hepatocytes) Culture medium (rat hepatocytes) | ─ galN ↑ LPS ↑ | [48] [51] |
| UGT2B3 | Protein | Culture medium (rat hepatocytes) | ─ | [48] |
| UGT2B4 | mRNA, protein | Plasma | ─ | [50,57] |
| UGT2B5 | Protein | Culture medium (rat hepatocytes) | ─ | [48] |
| UGT2B7 | mRNA, protein | Plasma | ─ | [50,57] |
| UGT2B10 | mRNA, protein | Plasma | ─ | [50,57] |
| UGT2B11 | mRNA | Plasma | ─ | [57] |
| UGT2B15 | mRNA, protein | Plasma | ─ | [50,57] |
| UGT2B17 | Protein mRNA | Culture medium (rat hepatocytes) Plasma | galN ↑ ─ | [51] [57] |
| UGT2B37 | Protein | Culture medium (rat hepatocytes) | galN ↑ | [51] |
| UGT3A1 | mRNA | Plasma | ─ | [57] |
| UGT3A2 | mRNA | Plasma | ─ | [57] |
| Uptake Transporter | Detection Level | Sample | References |
|---|---|---|---|
| OAT1 (SLC22A6) | Protein | Urine | [62] |
| OAT3 (SLC22A8) | Protein | Urine | [62] |
| OAT4 (SLC22A11) | Protein | Urine | [62] |
| OAT10 (SLC22A13) | Protein | Urine | [62] |
| OATP1A1 (SLCO1A1) | Protein | Urine (rat) | [65] |
| OATP1A3 (SLCO1A3) | Protein | Culture medium (rat hepatocytes) | [48] |
| OATP1A4 (SLCO1A4) | Protein | Culture medium (rat hepatocytes) | [48] |
| OATP1B1 (SLCO1B1) | mRNA, Protein | Plasma | [55,57] |
| OATP1B3 (SLCO1B3) | Protein | Plasma | [55] |
| OATP4C1 (SLCO4C1) | Protein | Urine | [62] |
| OCT2 (SLC22A2) | Protein | Urine | [62] |
| OCTN2 (SLC22A5) | Protein | Urine | [62] |
| Efflux Transporter | Detection Level | Sample | References |
|---|---|---|---|
| P-gp (ABCB1) | mRNA Protein | Plasma Urine | [57,58] [60,62] |
| MRP2 (ABCC2) | mRNA | Plasma | [57] |
| BCRP (ABCG2) | Regulatory miRNA * mRNA | Plasma Plasma | [68] [57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gagliardi, A.; Bajraktari-Sylejmani, G.; Barocelli, E.; Weiss, J.; Rigalli, J.P. Extracellular Vesicles as Surrogates for Drug Metabolism and Clearance: Promise vs. Reality. Life 2023, 13, 1745. https://doi.org/10.3390/life13081745
Gagliardi A, Bajraktari-Sylejmani G, Barocelli E, Weiss J, Rigalli JP. Extracellular Vesicles as Surrogates for Drug Metabolism and Clearance: Promise vs. Reality. Life. 2023; 13(8):1745. https://doi.org/10.3390/life13081745
Chicago/Turabian StyleGagliardi, Anna, Gzona Bajraktari-Sylejmani, Elisabetta Barocelli, Johanna Weiss, and Juan Pablo Rigalli. 2023. "Extracellular Vesicles as Surrogates for Drug Metabolism and Clearance: Promise vs. Reality" Life 13, no. 8: 1745. https://doi.org/10.3390/life13081745
APA StyleGagliardi, A., Bajraktari-Sylejmani, G., Barocelli, E., Weiss, J., & Rigalli, J. P. (2023). Extracellular Vesicles as Surrogates for Drug Metabolism and Clearance: Promise vs. Reality. Life, 13(8), 1745. https://doi.org/10.3390/life13081745