
Probe Capture Enrichment Methods for HIV and HCV Genome Sequencing and Drug Resistance Genotyping


Abstract
:1. Introduction
2. Overview of Experimental Methods
3. Extraction Method
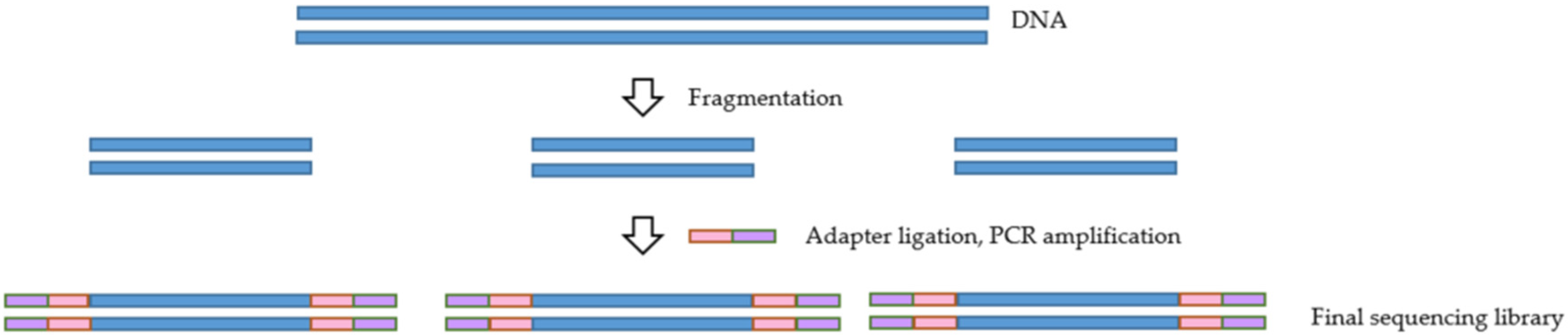
4. Library Preparation Method
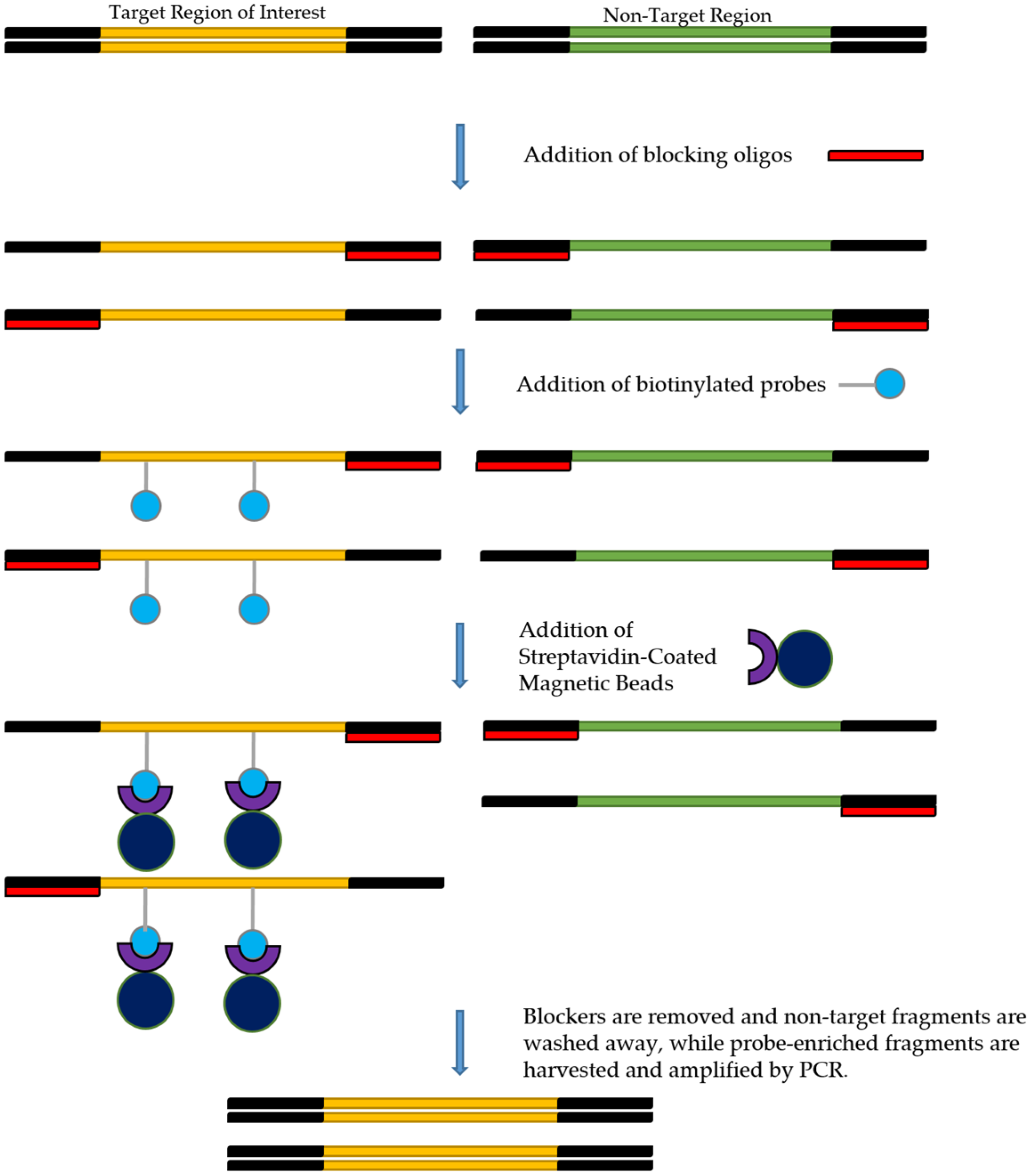
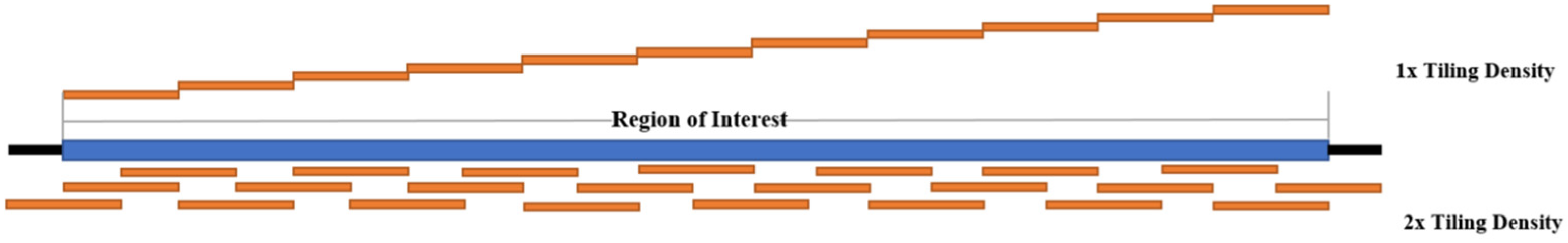
5. Target Enrichment
6. Next-Generation Sequencing
7. Post-Sequencing Analysis (Bioinformatics)
8. Target Enrichment Performance
9. Limitations
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- HIV/AIDS. UNAIDS Data 2020. 432 Geneva, Switzerland. 2020. Available online: https://www.unaids.org/en/resources/documents/2020/unaids-data (accessed on 25 March 2022).
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 9 March 2022).
- UNAIDS. Fast-Track- Ending the AIDs Epidemic by 2030. Available online: https://www.unaids.org/sites/default/files/media_asset/JC2686_WAD2014report_en.pdf (accessed on 10 March 2022).
- World Health Organisation. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 9 March 2022).
- Tough, R.H.; Tough, R.H.; McLaren, P.J.; McLaren, P.J.; Tough, R.H.; Tough, R.H.; McLaren, P.J.; McLaren, P.J. Interaction of the Host and Viral Genome and Their Influence on HIV Disease. Front. Genet. 2019, 9, 720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobran, S.T.; Ancuta, P.; Shoukry, N.H. A Tale of Two Viruses: Immunological Insights Into HCV/HIV Coinfection. Front. Immunol. 2021, 12, 726419. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.A.; Nevot, M.; Jordan-Paiz, A.; Franco, S. Similarities between Human Immunodeficiency Virus Type 1 and Hepatitis C Virus Genetic and Phenotypic Protease Quasispecies Diversity. J. Virol. 2015, 89, 9758–9764. [Google Scholar] [CrossRef] [Green Version]
- Manyana, S.; Gounder, L.; Pillay, M.; Manasa, J.; Naidoo, K.; Chimukangara, B. HIV-1 Drug Resistance Genotyping in Resource Limited Settings: Current and Future Perspectives in Sequencing Technologies. Viruses 2021, 13, 1125. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Hundie, G.B.; Schürch, A.; Smits, S.L.; Pas, S.D.; Le Pogam, S.; Janssen, H.L.A.; De Knegt, R.J.; Osterhaus, A.D.M.E.; Najera, I.; et al. Identification of HCV Resistant Variants against Direct Acting Antivirals in Plasma and Liver of Treatment Naïve Patients. Sci. Rep. 2017, 7, 4688. [Google Scholar] [CrossRef]
- Simen, B.; Simons, J.F.; Hullsiek, K.H.; Novak, R.M.; MacArthur, R.D.; Baxter, J.D.; Huang, C.; Lubeski, C.; Turenchalk, G.S.; Braverman, M.S.; et al. Low-Abundance Drug-Resistant Viral Variants in Chronically HIV-Infected, Antiretroviral Treatment–Naive Patients Significantly Impact Treatment Outcomes. J. Infect. Dis. 2009, 199, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Inzaule, S.C.; Ondoa, P.; Peter, T.; Mugyenyi, P.N.; Stevens, W.S.; de Wit, T.F.R.; Hamers, R.L. Affordable HIV drug-resistance testing for monitoring of antiretroviral therapy in sub-Saharan Africa. Lancet Infect. Dis. 2016, 16, e267–e275. [Google Scholar] [CrossRef]
- Masquelier, B. Low-Frequency HIV-1 Drug Resistance Mutations and Risk of NNRTI-Based Antiretroviral Treatment Failure. JAMA 2011, 305, 1327–1335. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, A.H.; Rupnik, A.; O’Shea, H.; Crispie, F.; Keaveney, S.; Cotter, P. High Throughput Sequencing for the Detection and Characterization of RNA Viruses. Front. Microbiol. 2021, 12, 621719. [Google Scholar] [CrossRef]
- Aitken, S.C.; Wallis, C.L.; Stevens, W.; de Wit, T.R.; Schuurman, R. Stability of HIV-1 Nucleic Acids in Dried Blood Spot Samples for HIV-1 Drug Resistance Genotyping. PLoS ONE 2015, 10, e0131541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudin, M.; Desnues, C. Hybrid Capture-Based Next Generation Sequencing and Its Application to Human Infectious Diseases. Front. Microbiol. 2018, 9, 2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, S.Y.; Bose, N.; Gonçalves, A.B.R.; Erlich, H.A.; Calloway, C.D. Applications of Probe Capture Enrichment Next Generation Sequencing for Whole Mitochondrial Genome and 426 Nuclear SNPs for Forensically Challenging Samples. Genes 2018, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonsall, D.; Golubchik, T.; De Cesare, M.; Limbada, M.; Kosloff, B.; MacIntyre-Cockett, G.; Hall, M.; Wymant, C.; Ansari, M.A.; Abeler-Dörner, L.; et al. A Comprehensive Genomics Solution for HIV Surveillance and Clinical Monitoring in Low-Income Settings. J. Clin. Microbiol. 2020, 58, e00382-20. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Olivo, A.; Laeyendecker, O.; Forberg, K.; Ndembi, N.; Mbanya, D.; Kaptue, L.; Quinn, T.C.; Cloherty, G.A.; Rodgers, M.A.; et al. Universal Target Capture of HIV Sequences from NGS Libraries. Front. Microbiol. 2018, 9, 2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnikov, A.; Galinsky, K.; Rogov, P.; Fennell, T.; Van Tyne, D.; Russ, C.; Daniels, R.; Barnes, K.G.; Bochicchio, J.; Ndiaye, D.; et al. Hybrid selection for sequencing pathogen genomes from clinical samples. Genome Biol. 2011, 12, R73. [Google Scholar] [CrossRef] [Green Version]
- Amorim-Vaz, S.; Tran, V.D.T.; Pradervand, S.; Pagni, M.; Coste, A.T.; Sanglard, D. RNA Enrichment Method for Quantitative Transcriptional Analysis of Pathogens In Vivo Applied to the Fungus Candida albicans. mBio 2015, 6, e00942-15. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.C.; Bryant, J.M.; Einer-Jensen, K.; Holdstock, J.; Houniet, D.T.; Chan, J.Z.M.; Depledge, D.P.; Nikolayevskyy, V.; Broda, A.; Stone, M.J.; et al. Rapid Whole-Genome Sequencing of Mycobacterium tuberculosis Isolates Directly from Clinical Samples. J. Clin. Microbiol. 2015, 53, 2230–2237. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, M.T.; Brown, A.C.; Kundu, S.; Tutill, H.J.; Williams, R.; Brown, J.R.; Holdstock, J.; Holland, M.J.; Stevenson, S.; Dave, J.; et al. Whole-genome enrichment and sequencing of Chlamydia trachomatisdirectly from clinical samples. BMC Infect. Dis. 2014, 14, 591. [Google Scholar] [CrossRef]
- Bonsall, D.; Ansari, M.A.; Ip, C.L.; Trebes, A.; Brown, A.; Klenerman, P.; Buck, D.S.; Piazza, P.; Barnes, E.; Bowden, R.; et al. ve-SEQ: Robust, unbiased enrichment for streamlined detection and whole-genome sequencing of HCV and other highly diverse pathogens. F1000Research 2015, 4, 1062. [Google Scholar] [CrossRef] [Green Version]
- Iwase, S.C.; Miyazato, P.; Katsuya, H.; Islam, S.; Yang, B.T.J.; Ito, J.; Matsuo, M.; Takeuchi, H.; Ishida, T.; Matsuda, K.; et al. HIV-1 DNA-capture-seq is a useful tool for the comprehensive characterization of HIV-1 provirus. Sci. Rep. 2019, 9, 12326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazato, P.; Katsuya, H.; Fukuda, A.; Uchiyama, Y.; Matsuo, M.; Tokunaga, M.; Hino, S.; Nakao, M.; Satou, Y. Application of targeted enrichment to next-generation sequencing of retroviruses integrated into the host human genome. Sci. Rep. 2016, 6, 28324. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, S.; Kirchner, R.; Amr, S.S.; Mansur, L.; Shakhbatyan, R.; Kim, M.; Bosque, A.; Siliciano, R.F.; Planelles, V.; Hofmann, O.; et al. HIV Integration Site Analysis of Cellular Models of HIV Latency with a Probe-Enriched Next-Generation Sequencing Assay. J. Virol. 2016, 90, 4511–4519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, E.; Vattipally, B.S.; Badhan, A.; Christiansen, M.T.; Adamson, W.; Ansari, M.A.; Bibby, D.; Breuer, J.; Brown, A.; Bowden, R.; et al. Comparison of Next-Generation Sequencing Technologies for Comprehensive Assessment of Full-Length Hepatitis C Viral Genomes. J. Clin. Microbiol. 2016, 54, 2470–2484. [Google Scholar] [CrossRef] [Green Version]
- Colson, P.; Dhiver, C.; Tamalet, C.; Delerce, J.; Glazunova, O.O.; Gaudin, M.; Levasseur, A.; Raoult, D. Dramatic HIV DNA degradation associated with spontaneous HIV suppression and disease-free outcome in a young seropositive woman following her infection. Sci. Rep. 2020, 10, 2548. [Google Scholar] [CrossRef]
- Charre, C.; Ginevra, C.; Sabatier, M.; Regue, H.; Destras, G.; Brun, S.; Burfin, G.; Scholtes, C.; Morfin, F.; Valette, M.; et al. Evaluation of NGS-based approaches for SARS-CoV-2 whole genome characterisation. Virus Evol. 2020, 6, veaa075. [Google Scholar] [CrossRef]
- Martínez-Puchol, S.; Itarte, M.; Rusiñol, M.; Forés, E.; Mejías-Molina, C.; Andrés, C.; Antón, A.; Quer, J.; Abril, J.F.; Girones, R.; et al. Exploring the diversity of coronavirus in sewage during COVID-19 pandemic: Don’t miss the forest for the trees. Sci. Total Environ. 2021, 800, 149562. [Google Scholar] [CrossRef]
- Berg, M.G.; Yamaguchi, J.; Alessandri-Gradt, E.; Tell, R.W.; Plantier, J.-C.; Brennan, C.A. A Pan-HIV Strategy for Complete Genome Sequencing. J. Clin. Microbiol. 2016, 54, 868–882. [Google Scholar] [CrossRef] [Green Version]
- Ali, N.; Rampazzo, R.; Costa, A.D.T.; Krieger, M.A. Current Nucleic Acid Extraction Methods and Their Implications to Point-of-Care Diagnostics. BioMed Res. Int. 2017, 2017, 9306564. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, M.; Gall, A.; Vink, M.; Zorgdrager, F.; Binter, Š.; Edwards, S.; Jurriaans, S.; Bakker, M.; Ong, S.H.; Gras, L.; et al. From clinical sample to complete genome: Comparing methods for the extraction of HIV-1 RNA for high-throughput deep sequencing. Virus Res. 2017, 239, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Guichet, E.; Serrano, L.; Laurent, C.; Eymard-Duvernay, S.; Kuaban, C.; Vidal, L.; Delaporte, E.; Ngole, E.M.; Ayouba, A.; Peeters, M. Comparison of different nucleic acid preparation methods to improve specific HIV-1 RNA isolation for viral load testing on dried blood spots. J. Virol. Methods 2017, 251, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Dhummakupt, A.; Siems, L.; Persaud, D. Alternative Sample Types for HIV-1 Antiretroviral Drug Resistance Testing. J. Infect. Dis. 2017, 216, S834–S837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dijk, E.L.; Jaszczyszyn, Y.; Thermes, C. Library preparation methods for next-generation sequencing: Tone down the bias. Exp. Cell Res. 2014, 322, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Integrated DNA Technologies IDT. Available online: https://www.idtdna.com (accessed on 18 January 2022).
- Agilent Technologies. Available online: https://www.agilent.com/cs/library/usermanuals/public/G7530-90000.pdf (accessed on 25 March 2022).
- Agilent Technologies. Available online: https://www.agilent.com/cs/library/usermanuals/Public/G9691-90000.pdf (accessed on 25 March 2022).
- Arbor Biosciences. Available online: https://arborbiosci.com/genomics/targeted-sequencing/mybaits/ (accessed on 11 March 2022).
- Integrated DNA Technologies IDT. Available online: https://www.idtdna.com/pages/products/next-generation-sequencing/workflow/xgen-ngs-hybridization-capture?utm_source=google&utm_medium=cpc&utm_campaign=ga_ngs&utm_content=ad_group_hyb_capture&gclid=EAIaIQobChMI3feYsL7i9gIVshbUAR1G9AFyEAAYASAAEgJrsvD_BwE (accessed on 18 January 2022).
- Lucigen. Available online: https://www.lucigen.com/nxseq-hybcap-target-enrichment-kit/ (accessed on 11 March 2022).
- Roche. Available online: https://www.roche.com/ (accessed on 11 March 2022).
- Hale, H.; Gardner, E.M.; Viruel, J.; Pokorny, L.; Johnson, M.G. Strategies for reducing per-sample costs in target capture sequencing for phylogenomics and population genomics in plants. Appl. Plant Sci. 2020, 8, e11337. [Google Scholar] [CrossRef] [Green Version]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Samples Tested | Extraction Method | Ref |
|---|---|---|---|
| HIV | HIV-1 (plasma) | m2000sp RNA protocol (Abbott Laboratories) | [19] |
| HIV | HIV-1 (subtype B) from HIV-1-infected latent cell lines | DNeasy Blood and Tissue Kit (Qiagen) | [25] |
| HIV | HIV-1-infected cell line (ACH-2) | DNeasy Blood and Tissue Kit (Qiagen) | [26] |
| HCV | Clinical HCV samples (plasma) | NucliSENS Magnetic Extraction System (bioMérieux) | [24] |
| HIV | HIV-1 infected cell lines (ACH-2, J-Lat) | Gentra Puregene cell kit (Qiagen) | [27] |
| HCV | Clinical HCV samples (plasma), In vitro RNA transcipts, assay controls (plasma) | Agencourt RNAdvance blood kit (Beckman Coulter), QIAamp viral RNA minikit (Qiagen), NucliSENS magnetic extration system (bioMérieux) | [28] |
| HIV | Clinical HIV samples (plasma) | NucliSENS easyMAG system (bioMérieux) | [18] |
| HIV | Clinical HIV samples from peripheral blood mononuclear cells | EZ1 Virus Mini Kit v2.0 (Qiagen) | [29] |
| Virus | Library Preparation Method | Ref |
|---|---|---|
| HIV | Nextera XT Kit (Illumina) | [19,29] |
| HIV | NEBNext UltraDNA II library preparation kit and NEBNExt multiplex oligos for Illumina (New England BioLabs) | [25,26] |
| HCV | NEBNext® UltraTM Directional RNA Library Prep Kit for Illumina® (New England Biolabs) | [24] |
| HIV | SPRI-TE nucleic acid extractor automated library preparation (Beckman Coulter) with NEXTflex adapters (Bioo Scientific) | [27] |
| HCV | KAPA Library Preparation Kit with index tagging using KAPA HiFi HotStart (KAPA Biosystems) and NEBNext multiplex oligos for Illumina Index Primer Sets 1 and 2 (New England BioLabs), SureSelectXT Target Enrichment (Aligent), NEBNext Ultra Directional RNA Library Prep kit for Illumina (New England BioLabs) | [28] |
| HIV | SMARTer Stranded Total RNA-Seq Kit V2—Pico Input Mammalian (Clontech, Takara Bio) | [18] |
| Company | Kit | Compatible NGS Platforms | Type of Baits | Ref |
|---|---|---|---|---|
| Agilent Technologies | SureSelectXT Target Enrichment System | HiSeq, MiSeq, NextSeq 500, NovaSeq 6000 | Pre-designed or custom designed DNA probes | [40] |
| Agilent Technologies | SureSelectXT RNA Target Enrichment System | HiSeq, MiSeq, NextSeq 500 | Pre-designed or custom designed RNA probes | [41] |
| Arbor Biosciences | myBaits Hybridization Capture for Targeted NGS | Illumina platforms, Ion Torrent, PacBio, Oxford Nanopore Technologies | Pre-designed or custom designed RNA or DNA probes | [42] |
| Integrated DNA Technologies (IDT) | xGen™ NGS Hybridization Capture | Illumina platforms | Pre-designed or custom designed DNA probes | [43] |
| Lucigen | NxSeq HybCap Target Enrichment Kit | Illumina platforms, Ion Torrent | Custom designed RNA probes | [44] |
| Roche | NimbleGen Seq Cap EZ system | Illumina platforms | Pre-designed or custom designed DNA probes | [45] |
| Virus | Probe Design/Enrichment Method | Sequencing Platform | Ref |
|---|---|---|---|
| HIV | 120 nt biotinylated DNA probes based on consensus sequences of HIV-1 and HIV-2 (xGen Lockdown probes and reagents, Integrated DNA Technologies) | MiSeq (Illumina) | [19] |
| HIV | 120 nt biotinylated DNA probes based on HXB2 reference sequence (xGen Lockdown probes and reagents, Integrated DNA Technologies) | MiSeq or NextSeq (Illumina) | [25] |
| HIV | 120 nt biotinylated DNA probes based on HXB2 reference sequence (xGen Lockdown probes, Integrated DNA Technologies) with SeqCap EZ Hybridization and Wash Kit (Roche NimbleGen) | MiSeq or NextSeq (Illumina) | [26] |
| HCV | 120 nt DNA oligonucleotide probes (xGen Lockdown probes, Integrated DNA Technologies) and xGen® Lockdown® protocol (Integrated DNA Technologies) | MiSeq (Illumina) | [24] |
| HIV | 120 nt DNA oligonucleotide probes (xGen Lockdown probes, Integrated DNA Technologies) with Dynabeads MyOne Streptavidin T1 (Life Technologies), PCR enrichment with Kapa HiFi DNA polymerase | MiSeq (Illumina) | [27] |
| HCV | 120 nt RNA probes spanning 953 GenBank HCV reference genomes. Enrichment using xGen Lockdown protocol (Integrated DNA Technologies), NimbleGen Seq Cap EZ system (Roche), SureSelect Target Enrichment System (Agilent), or SureSelectXT Target Enrichment (Agilent) | MiSeq (Illumina) | [28] |
| HIV | Custom HIV-specific biotinylated 120 nt probe set (XGen Lockdown Probes, Integrated DNA Technologies) with SeqCap EZ hybridization and wash kit (Roche) | MiSeq (Illumina) | [18] |
| HIV | Custom HIV-specific 120 nt probes (Arbor Biosciences) used with the myBaits target capture kit (Arbor Biosceinces) | MiSeq (Illumina) | [29] |
| Virus | Bioinformatic Platforms Used | Ref |
|---|---|---|
| HIV | CLC Genomics Workbench 9.0 (CLC Bio) for analysis of reads, phylogenetic analysis using SIMPLOT | [19] |
| HIV | In-house Pearl script for selection of paired-reads and cleaning of the reads, BWA-MEM algorithm for alignment to reference, Samtools program and Picard command line tools to remove multiply aligned reads and duplicates, final aligned files visualized with Integrative Genomics Viewer (IGV) | [25] |
| HIV | BWA-MEM algorithm for mapping, Picard tool for the removal of PCR duplicates, Strand NGS (Strand Life Science) for the visualization of mapped data, Low Frequency Variant Detection Tool (CLC Genomics Workbench 7.5 software, CLC Bio) for error correction | [26] |
| HCV | QUASR v7.01 & CutAdapt v1.7.1 for trimming sequences, Bowtie v2.2.4 for comparision to human reference, BLASTn database for screenning reads, Vicuna v1.3 & V-FAT v1.0 for de novo assembly, Mosaik v2.2.28 for mapping reads back to assembly, V-Phaser v2.0 for calling variants, V-Profiler v1.0 for examining intra-host diversity | [24] |
| HIV | BWA-MEM algorithm for alignment, sambamba for marking duplicate alignments, Gene SeT AnaLysis Toolkit for gene ontology analysis | [27] |
| HCV | FastQC, Tanoti, in-house resistance mutation tools, de novo assembly using MetAmos Genome mapping, assembly, and finishing using CLC Genomics Workbench, DAA analysis using in-house script QUASR v7.01 & CutAdapt v1.7.1 for trimming sequences, Bowtie v2.2.4 for comparision to human reference, BLASTn database for screenning reads, Vicuna v1.3 & V-FAT v1.0 for de novo assembly, Mosaik v2.2.28 for mapping reads back to assembly, V-Phaser v2.0 for calling variants, V-Profiler v1.0 for examining intra-host diversity | [28] |
| HIV | Kraken for processing raw sequences, Trimmomatic for trimming sequences, SPAdes, metaSPAdes for assembly into contigs, cd-hit-est for cluster generation, shiver for mapping reads, Kallisto for mapping reads with no contigs assembled, phyloscanner for identifying and removing contaminant reads, Stanford drug resistance tool for determining consensus and minority drug resisitance levels | [18] |
| HIV | CLC Genomics Workbench software (CLC Bio/Qiagen) for analysis of reads and assembly by mapping to the HIV genome (HIV-1 Strain HXB2) from GenBank | [29] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munyuza, C.; Ji, H.; Lee, E.R. Probe Capture Enrichment Methods for HIV and HCV Genome Sequencing and Drug Resistance Genotyping. Pathogens 2022, 11, 693. https://doi.org/10.3390/pathogens11060693
Munyuza C, Ji H, Lee ER. Probe Capture Enrichment Methods for HIV and HCV Genome Sequencing and Drug Resistance Genotyping. Pathogens. 2022; 11(6):693. https://doi.org/10.3390/pathogens11060693
Chicago/Turabian StyleMunyuza, Chantal, Hezhao Ji, and Emma R. Lee. 2022. "Probe Capture Enrichment Methods for HIV and HCV Genome Sequencing and Drug Resistance Genotyping" Pathogens 11, no. 6: 693. https://doi.org/10.3390/pathogens11060693
APA StyleMunyuza, C., Ji, H., & Lee, E. R. (2022). Probe Capture Enrichment Methods for HIV and HCV Genome Sequencing and Drug Resistance Genotyping. Pathogens, 11(6), 693. https://doi.org/10.3390/pathogens11060693