

Exploration of the Nuclear Proteomes in the Ciliate Oxytricha trifallax
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Nuclear Enrichment
2.3. Mass Spectrometry of Separated Nuclear Samples
2.4. Analysis of Nuclear Mass Spectrometry Results
2.5. Quantitative Mass Spectrometry Analysis of Oxytricha Histone PTMs
2.6. Protein Predictions
2.7. Generation of Injection Constructs
2.8. Transformation
2.9. Western Blotting
2.10. Generation of Custom Antibodies
2.11. Cell Imaging
3. Results
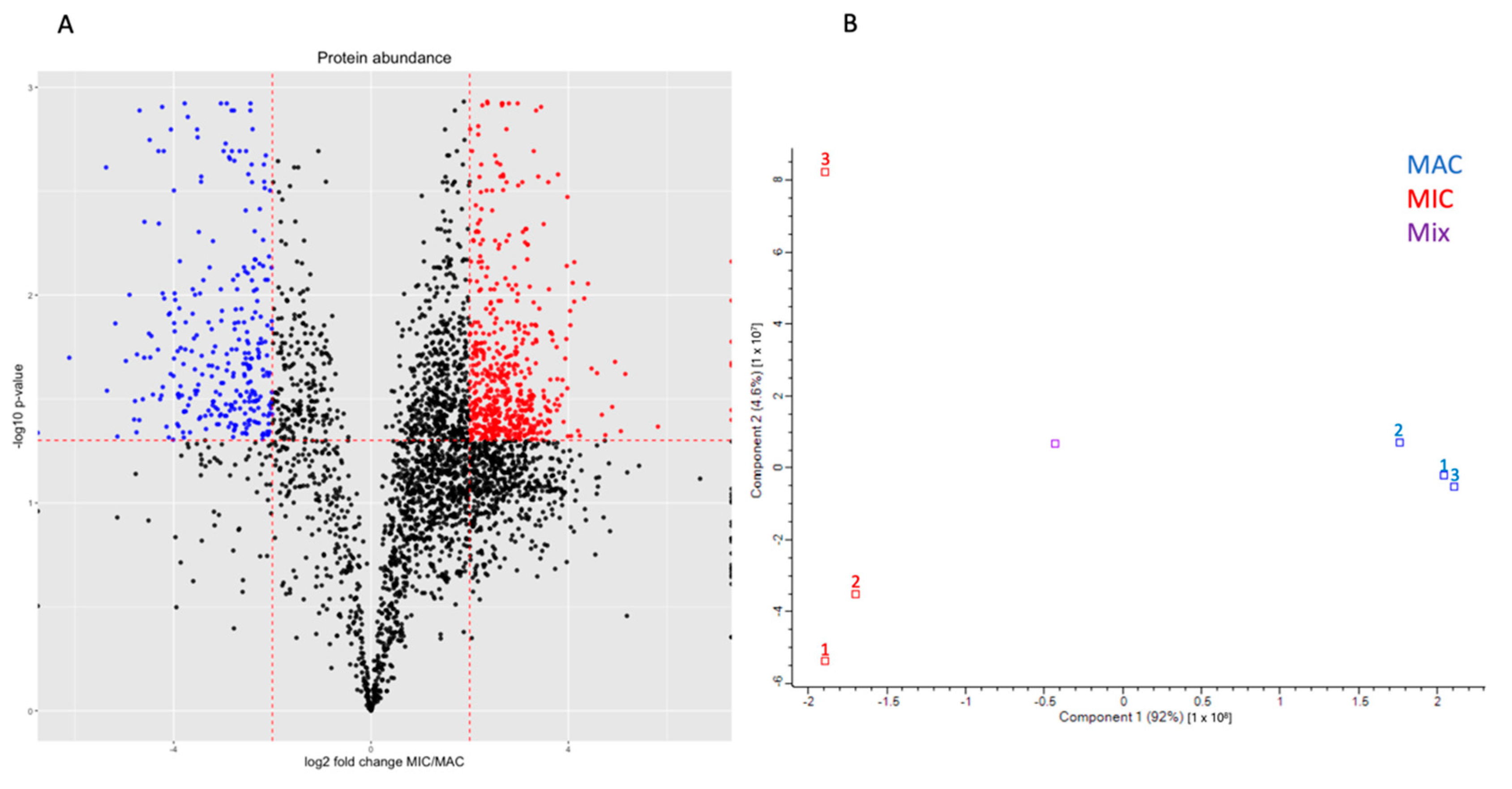
3.1. Oxytricha Nuclei Contain Distinct Proteomes
3.2. Micronuclear Fraction Also Includes Other Organelles
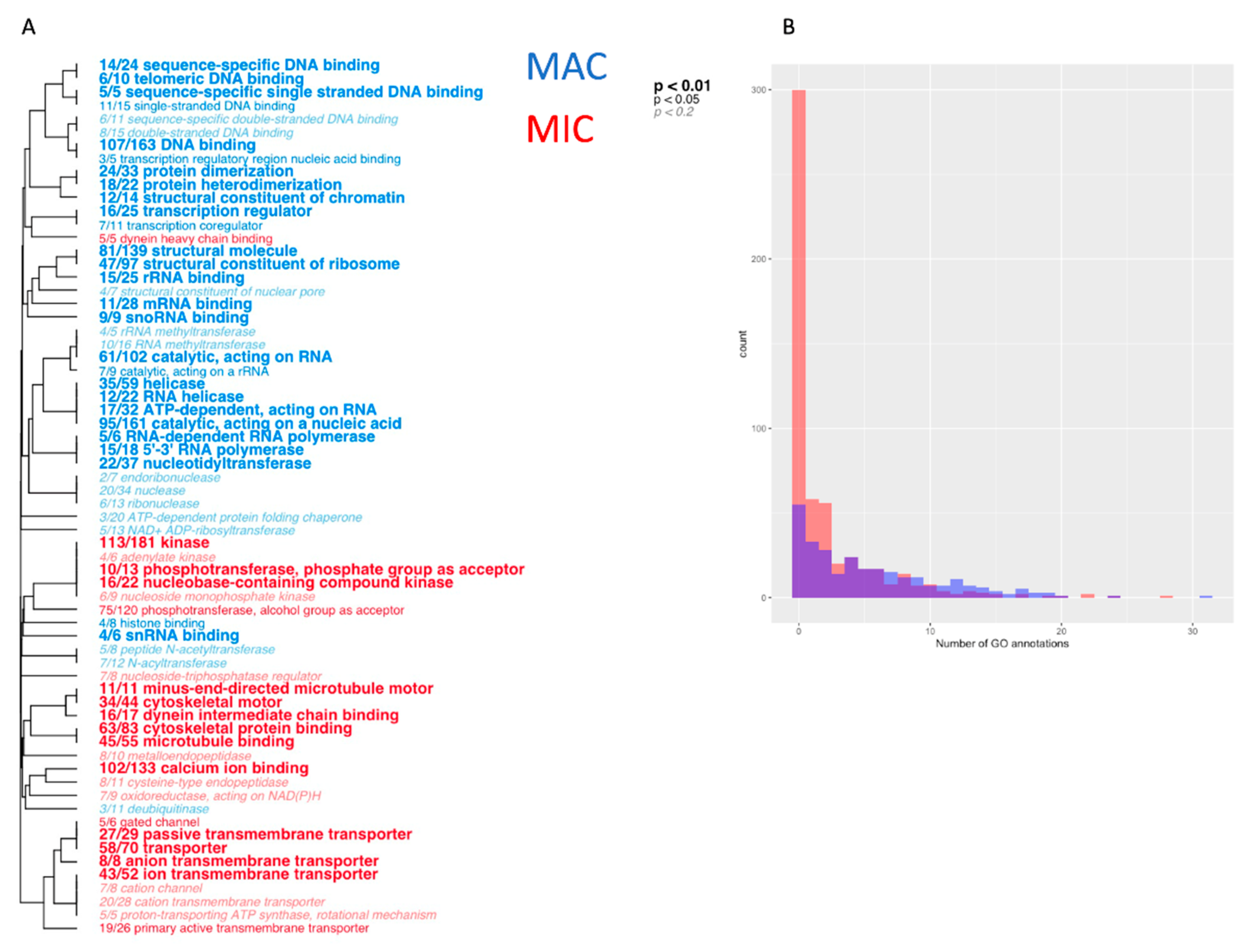
3.3. Distinct Functional Differences between MAC and MIC Proteins
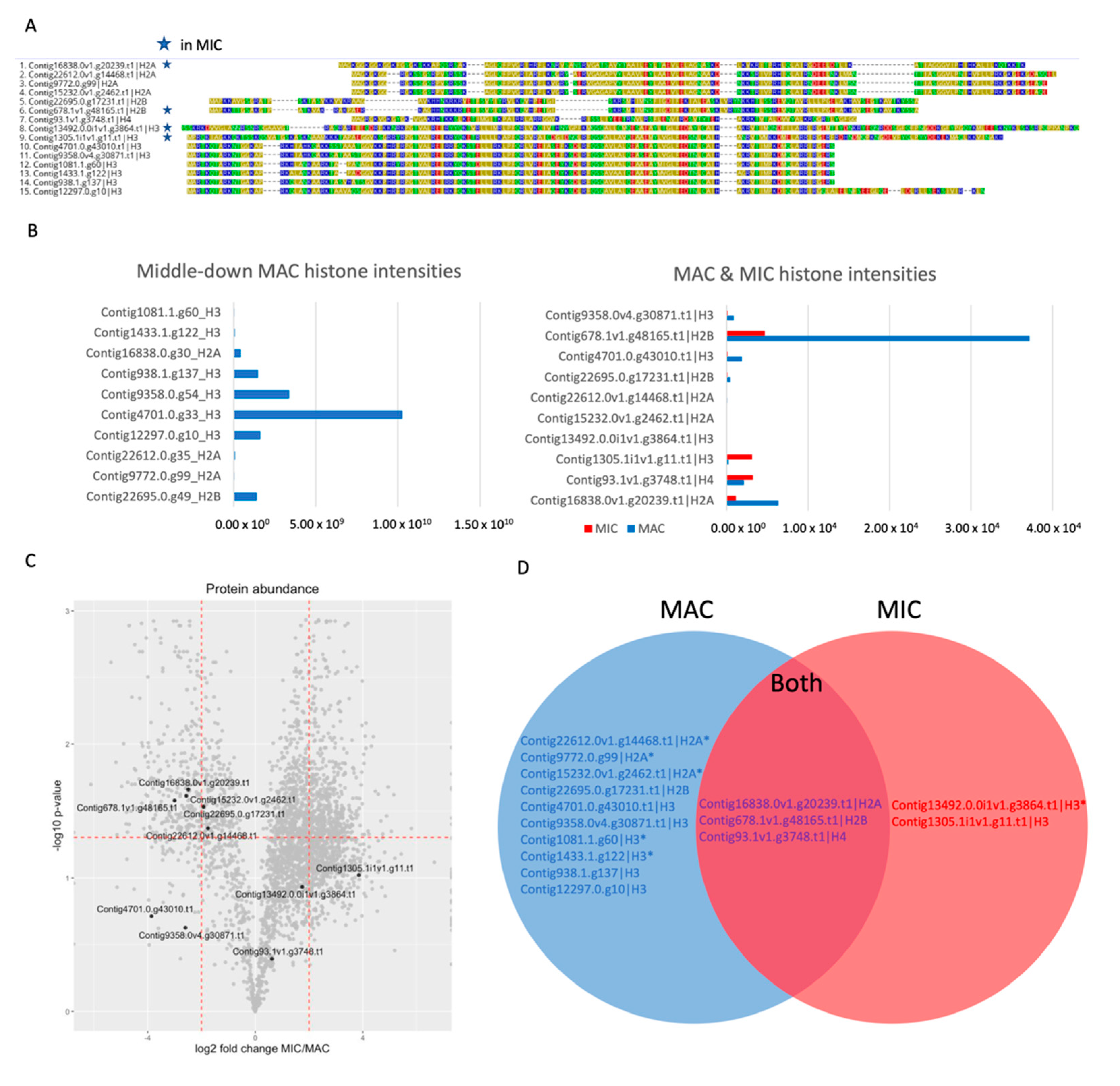
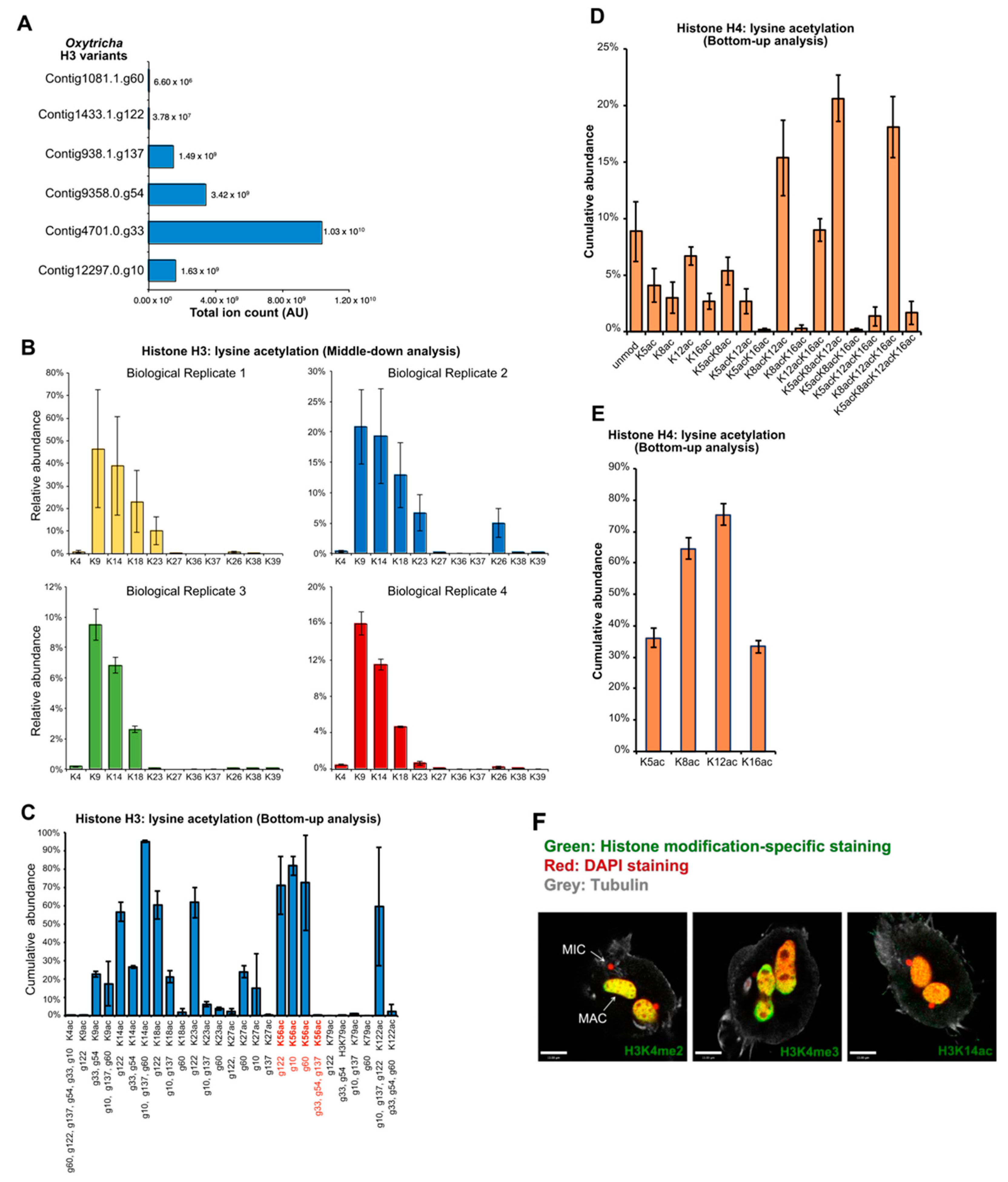
3.4. Different Sets of Core Histones Are Present in Respective Nuclei
3.5. MAC Histones Are Modified with Active Marks
3.6. RNA Polymerase Localizes Exclusively to the MAC
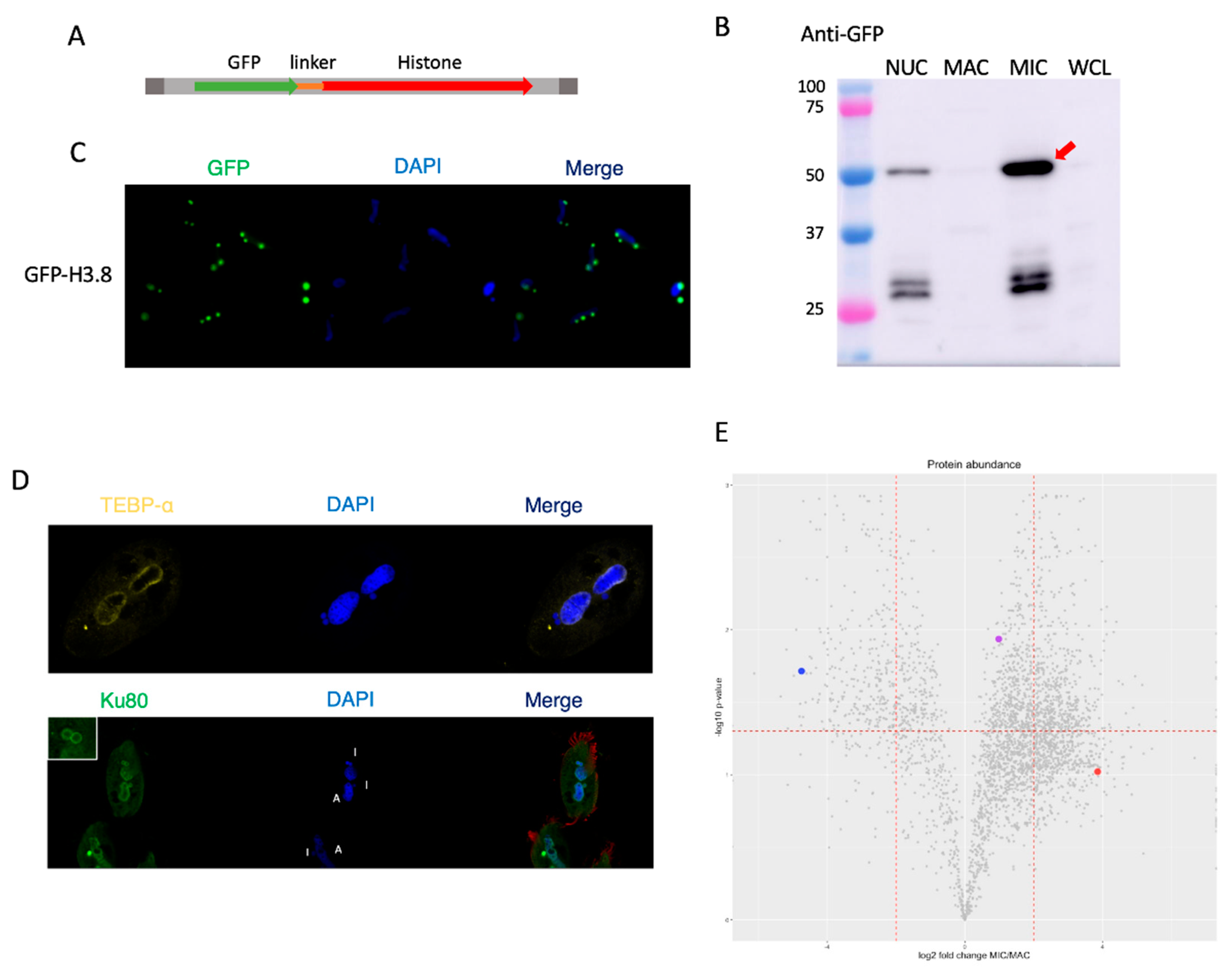
3.7. Histone H3 Variant H3.8 Exclusively Localizes to the MIC
3.8. Telomere End-Binding Protein Alpha Is Exclusively Localized to the MAC
3.9. Ku80 Is Present within Both Nuclei
3.10. MIC-Enriched Domains
3.11. MAC-Enriched Domains
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lamond, A.I.; Earnshaw, W.C. Structure and function in the nucleus. Science 1998, 280, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Bhat, P.; Honson, D.; Guttman, M. Nuclear compartmentalization as a mechanism of quantitative control of gene expression. Nat. Rev. Mol. Cell. Biol. 2021, 22, 653–670. [Google Scholar] [CrossRef]
- De Magistris, P.; Antonin, W. The Dynamic Nature of the Nuclear Envelope. Curr. Biol. 2018, 28, R487–R497. [Google Scholar] [CrossRef] [Green Version]
- Prescott, D.M. The DNA of ciliated protozoa. Microbiol. Rev. 1994, 58, 233–267. [Google Scholar] [CrossRef]
- Bracht, J.R.; Fang, W.; Goldman, A.D.; Dolzhenko, E.; Stein, E.M.; Landweber, L.F. Genomes on the edge: Programmed genome instability in ciliates. Cell 2013, 152, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Swart, E.C.; Bracht, J.R.; Magrini, V.; Minx, P.; Chen, X.; Zhou, Y.; Khurana, J.S.; Goldman, A.D.; Nowacki, M.; Schotanus, K.; et al. The Oxytricha trifallax macronuclear genome: A complex eukaryotic genome with 16,000 tiny chromosomes. PLoS Biol. 2013, 11, e1001473. [Google Scholar] [CrossRef] [Green Version]
- Lindblad, K.A.; Pathmanathan, J.S.; Moreira, S.; Bracht, J.R.; Sebra, R.P.; Hutton, E.R.; Landweber, L.F. Capture of complete ciliate chromosomes in single sequencing reads reveals widespread chromosome isoforms. BMC Genom. 2019, 20, 1037. [Google Scholar] [CrossRef] [Green Version]
- Caplan, E.B. Histones and other basic nuclear proteins in genetically active and genetically inactive nuclei of the ciliate, Oxytricha sp. Biochim. Biophys. Acta 1977, 479, 214–219. [Google Scholar] [CrossRef]
- Zhang, C.; Gao, S.; Molascon, A.J.; Liu, Y.; Andrews, P.C. Quantitative proteomics reveals histone modifications in crosstalk with H3 lysine 27 methylation. Mol. Cell. Proteom. 2014, 13, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, M.; Osakada, H.; Mori, C.; Fukuda, Y.; Nagao, K.; Obuse, C.; Hiraoka, Y.; Haraguchi, T. Compositionally distinct nuclear pore complexes of functionally distinct dimorphic nuclei in the ciliate Tetrahymena. J. Cell Sci. 2017, 130, 1822–1834. [Google Scholar] [CrossRef]
- Malone, C.D.; Falkowska, K.A.; Li, A.Y.; Galanti, S.E.; Kanuru, R.C.; Lamont, E.G.; Mazzarella, K.C.; Micev, A.J.; Osman, M.M.; Piotrowski, N.K.; et al. Nucleus-specific importin alpha proteins and nucleoporins regulate protein import and nuclear division in the binucleate Tetrahymena thermophila. Eukaryot. Cell 2008, 7, 1487–1499. [Google Scholar] [CrossRef] [Green Version]
- Beh, L.Y.; Debelouchina, G.T.; Clay, D.M.; Thompson, R.E.; Lindblad, K.A.; Hutton, E.R.; Bracht, J.R.; Sebra, R.P.; Muir, T.W.; Landweber, L.F. Identification of a DNA N6-Adenine Methyltransferase Complex and Its Impact on Chromatin Organization. Cell 2019, 177, 1781–1796.e25. [Google Scholar] [CrossRef]
- Lauth, M.R.; Spear, B.B.; Heumann, J.; Prescott, D.M. DNA of ciliated protozoa: DNA sequence diminution during macronuclear development of Oxytricha. Cell 1976, 7, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Perea, J.; Yu, Q.; Gygi, S.P.; Paulo, J.A. Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J. Proteome Res. 2018, 17, 2226–2236. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- R: A Language and Environment for Statistical Computing; R Core Team; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 18 May 2021).
- RStudio: Integrated Development for R; RStudio Team; RStudio, PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com/ (accessed on 1 July 2022).
- Yuan, Z.; Lin, S.; Molden, R.C.; Cao, X.; Bhanu, N.V.; Wang, X.; Sidoli, S.; Liu, S.; Garcia, B.A. EpiProfile Quantifies Histone Peptides with Modifications by Extracting Retention Time and Intensity in High-resolution Mass Spectra. Mol. Cell. Proteom. 2015, 14, 1696–1707. [Google Scholar] [CrossRef] [Green Version]
- Sidoli, S.; Schwämmle, V.; Ruminowicz, C.; Hansen, T.A.; Wu, X.; Helin, K.; Jensen, O.N. Middle-down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post-translational modifications in histones. Proteomics 2014, 14, 2200–2211. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef] [Green Version]
- Thumuluri, V.; Armenteros, J.J.A.; Johansen, A.R.; Nielsen, H.; Winther, O. DeepLoc 2.0: Multi-label subcellular localization prediction using protein language models. Nucleic Acids Res. 2022, 50, W228–W234. [Google Scholar] [CrossRef]
- Wright, R.M.; Aglyamova, G.V.; Meyer, E.; Matz, M.V. Gene expression associated with white syndromes in a reef building coral, Acropora hyacinthus. BMC Genom. 2015, 16, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2008, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, M.P.; Schweiker, V.L.; Bevilacqua, J.M.; Ruggles, J.A.; Schultz, S.C. Crystal structure of the Oxytricha nova telomere end binding protein complexed with single strand DNA. Cell 1998, 95, 963–974. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Wang, X.; Bracht, J.R.; Nowacki, M.; Landweber, L.F. Piwi-interacting RNAs protect DNA against loss during Oxytricha genome rearrangement. Cell 2012, 151, 1243–1255. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Bracht, J.R.; Goldman, A.D.; Dolzhenko, E.; Clay, D.M.; Swart, E.C.; Perlman, D.H.; Doak, T.G.; Stuart, A.; Amemiya, C.T.; et al. The Architecture of a Scrambled Genome Reveals Massive Levels of Genomic Rearrangement during Development. Cell 2014, 158, 1187–1198. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Neme, R.; Beh, L.Y.; Chen, X.; Braun, J.; Lu, M.W.; Landweber, L.F. Comparative genomics reveals insight into the evolutionary origin of massively scrambled genomes. eLife 2022, 11, e82979. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 30 November 2022).
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Smith, D.G.S.; Gawryluk, R.M.R.; Spencer, D.F.; Pearlman, R.E.; Siu, K.W.M.; Gray, M.W. Exploring the Mitochondrial Proteome of the Ciliate Protozoon Tetrahymena thermophila: Direct Analysis by Tandem Mass Spectrometry. J. Mol. Biol. 2007, 374, 837–863. [Google Scholar] [CrossRef]
- Swart, E.C.; Nowacki, M.; Shum, J.; Stiles, H.; Higgins, B.P.; Doak, T.G.; Schonatus, K.; Magrini, V.J.; Minx, P.; Mardis, E.R.; et al. The Oxytricha trifallax mitochondrial genome. Genome Biol. Evol. 2012, 4, 136–154. [Google Scholar] [CrossRef] [Green Version]
- Brunk, C.F.; Lee, L.C.; Tran, A.B.; Li, J. Complete sequence of the mitochondrial genome of Tetrahymena thermophila and comparative methods for identifying highly divergent genes. Nucleic Acids Res. 2003, 31, 1673–1682. [Google Scholar] [CrossRef]
- Guérin, F.; Arnaiz, O.; Boggetto, N.; Wilkes, C.D.; Meyer, E.; Sperling, L.; Duharcourt, S. Flow cytometry sorting of nuclei enables the first global characterization of Paramecium germline DNA and transposable elements. BMC Genom. 2017, 18, 327. [Google Scholar] [CrossRef] [Green Version]
- Zangarelli, C.; Arnaiz, O.; Bourge, M.; Gorrichon, K.; Jaszczyszyn, Y.; Mathy, N.; Escoriza, L.; Betermier, M.; Regnier, V. Developmental timing of programmed DNA elimination in Paramecium tetraurelia recapitulates germline transposon evolutionary dynamics. Genome Res. 2022, 11–12, 2028–2042. [Google Scholar] [CrossRef]
- Schlegel, M.; Muller, S.; Ruder, F.; Büsen, W. Transcriptionally inactive micronuclei, macronuclear anlagen and transcriptionally active macronuclei differ in histone composition in the hypotrichous ciliate Stylonychia lemnae. Chromosoma 1990, 99, 401–406. [Google Scholar] [CrossRef]
- Butler, A.P.; Laughlin, T.J.; Cadilla, C.L.; Henry, J.M.; Olins, D.E. Physical structure of gene-sized chromatin from the protozoan Oxytricha. Nucleic Acids Res. 1984, 12, 3201–3217. [Google Scholar] [CrossRef] [Green Version]
- Aeschlimann, S.H.; Jönsson, F.; Postberg, J.; Stover, N.A.; Petera, R.L.; Lipps, H.J.; Nowacki, M.; Swart, E.C. The Draft Assembly of the Radically Organized Stylonychia lemnae Macronuclear Genome. Genome Biol. Evol. 2014, 6, 1707–1723. [Google Scholar] [CrossRef]
- Nabeel-Shah, S.; Ashraf, K.; Saettone, A.; Garg, J.; Derynck, J.; Lambert, J.; Pearlman, R.E.; Fillingham, J. Nucleus-specific linker histones Hho1 and Mlh1 form distinct protein interactions during growth, starvation and development in Tetrahymena thermophila. Sci. Rep. 2020, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Postberg, J.; Heyse, K.; Cremer, M.; Cremer, T.; Lipps, H.J. Spatial and temporal plasticity of chromatin during programmed DNA-reorganization in Stylonychia macronuclear development. Epigenetics Chromatin 2008, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Grau-Bové, X.; Navarrete, C.; Chiva, C.; Pribasnig, T.; Antó, M.; Torruella, G.; Galindo, L.J.; Lang, B.F.; Moreira, D.; López-Garcia, P.; et al. A phylogenetic and proteomic reconstruction of eukaryotic chromatin evolution. Nat. Ecol. Evol. 2022, 6, 1007–1023. [Google Scholar] [CrossRef]
- Wahab, S.; Saettone, A.; Nabeel-Shah, S.; Dannah, N.; Fillingham, J. Exploring the Histone Acetylation Cycle in the Protozoan Model Tetrahymena thermophila. Front. Cell Dev. Biol. 2020, 8, 509. [Google Scholar] [CrossRef]
- Khurana, J.S.; Wang, X.; Chen, X.; Perlman, D.H.; Landweber, L.F. Transcription-independent functions of an RNA polymerase II subunit, Rpb2, during genome rearrangement in the ciliate, Oxytricha trifallax. Genetics 2014, 197, 839–849. [Google Scholar] [CrossRef]
- Forcob, S.; Bulic, A.; Jönsson, F.; Lipps, H.J.; Postberg, J. Differential expression of histone H3 genes and selective association of the variant H3.7 with a specific sequence class in Stylonychia macronuclear development. Epigenetics Chromatin 2014, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Gottschling, D.E.; Zakian, V.A. Telomere proteins: Specific recognition and protection of the natural termini of Oxytricha macronuclear DNA. Cell 1986, 47, 195–205. [Google Scholar] [CrossRef]
- Fang, G.; Cech, T.R. Oxytricha telomere-binding protein: DNA-dependent dimerization of the alpha and beta subunits. Proc. Natl. Acad. Sci. USA 1993, 90, 6056–6060. [Google Scholar] [CrossRef] [Green Version]
- Klobutcher, L.A.; Swanton, M.T.; Donini, P.; Prescott, D.M. All gene-sized DNA molecules in four species of hypotrichs have the same terminal sequence and an unusual 3’ terminus. Proc. Natl. Acad. Sci. USA 1981, 78, 3015–3019. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef]
- Fisher, T.S.; Zakian, V.A. Ku: A multifunctional protein involved in telomere maintenance. DNA Repair 2005, 4, 1215–1226. [Google Scholar] [CrossRef]
- Lin, I.T.; Chao, J.L.; Yao, M.C. An essential role for the DNA breakage-repair protein Ku80 in programmed DNA rearrangements in Tetrahymena thermophila. Mol. Biol. Cell 2012, 23, 2213–2225. [Google Scholar] [CrossRef]
- Abello, A.; Régnier, V.; Arnaiz, O.; Le Bars, R.; Bétermier, M.; Bischerour, J. Functional diversification of Paramecium Ku80 paralogs safeguards genome integrity during precise programmed DNA elimination. PLoS Genet. 2020, 16, e1008723. [Google Scholar] [CrossRef] [Green Version]
- Kilburn, C.L.; Pearson, C.G.; Romijn, E.P.; Meehl, J.B.; Giddings Jr, T.H.; Culver, B.P.; Yates, J.R., 3rd; Winey, M. New Tetrahymena basal body protein components identify basal body domain structure. J. Cell Biol. 2007, 178, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Longo, L.M.; Jablonska, J.; Vyas, P.; Kanade, M.; Kolodny, R.; Ben-Tal, N.; Tawfik, D.S. On the emergence of P-Loop NTPase and Rossmann enzymes from a Beta-Alpha-Beta ancestral fragment. eLife 2020, 9, e64415. [Google Scholar] [CrossRef]
- Bischoff, F.R.; Ponstingl, H. Mitotic regulator protein RCC1 is complexed with a nuclear ras-related polypeptide. Proc. Natl. Acad. Sci. USA 1991, 88, 10830–10834. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.K.; Barton, M.C. Bromodomain Histone Readers and Cancer. J. Mol. Biol. 2017, 429, 2003–2010. [Google Scholar] [CrossRef]
- Iwamoto, M.; Mori, C.; Osakada, H.; Koujin, T.; Hiraoka, Y.; Haraguchi, T. Nuclear localization signal targeting to macronucleus and micronucleus in binucleated ciliate Tetrahymena thermophila. Genes Cells 2018, 23, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Xu, J.; Bo, T.; Wang, W. Micronucleus-specific histone H1 is required for micronuclear chromosome integrity in Tetrahymena thermophila. PLoS ONE 2017, 12, e0187475. [Google Scholar] [CrossRef] [Green Version]
- Cervantes, M.D.; Xi, X.; Vermaak, D.; Yao, M.C.; Malik, H.S. The CNA1 Histone of the Ciliate Tetrahymena thermophila is Essential for Chromosome Segregation in the Germline Micronucleus. Mol. Biol. Cell 2006, 17, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Lhuillier-Akakpo, M.; Guérin, F.; Frapporti, A.; Duharcourt, S. DNA deletion as a mechanism for developmentally programmed centromere loss. Nucleic Acids Res. 2016, 44, 1553–1565. [Google Scholar] [CrossRef] [Green Version]
- Dawson, D.; Herrick, G. Telomeric properties of C4A4-homologous sequences in micronuclear DNA of Oxytricha fallax. Cell 1984, 36, 171–177. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| InterPro Domain | Count |
|---|---|
| EF-hand domain pair | 36 |
| WD40/YVTN repeat-like-containing domain superfamily | 26 |
| P-loop containing nucleoside triphosphate hydrolase | 21 |
| Leucine-rich repeat domain superfamily | 17 |
| Armadillo-type fold | 13 |
| Tetratricopeptide-like helical domain superfamily | 13 |
| Protein kinase-like domain superfamily | 10 |
| WD40 repeat, conserved site | 10 |
| Regulator of chromosome condensation, RCC1 | 9 |
| C2 domain superfamily | 6 |
| GAR domain | 6 |
| Giardin subunit beta-like | 6 |
| Growth factor receptor cysteine-rich domain superfamily | 6 |
| Kinesin motor domain | 6 |
| Serine/threonine-protein kinase, active site | 6 |
| Transient receptor potential cation channel subfamily V | 6 |
| Band 7 domain | 5 |
| Ion transport domain | 5 |
| IQ motif, EF-hand binding site | 5 |
| Papain-like cysteine peptidase superfamily | 5 |
| Protein kinase, ATP-binding site | 5 |
| Quinoprotein alcohol dehydrogenase-like superfamily | 5 |
| 14-3-3 domain | 4 |
| AAA+ ATPase domain | 4 |
| Ankyrin repeat-containing domain superfamily | 4 |
| B-box-type zinc finger | 4 |
| Enkurin domain | 4 |
| Kinesin-like protein | 4 |
| MORN motif | 4 |
| SAS-6, N-terminal domain superfamily | 4 |
| Zinc finger, RING/FYVE/PHD-type | 4 |
| InterPro Domain | Count |
|---|---|
| WD40 repeat-containing domain superfamily | 28 |
| P-loop containing nucleoside triphosphate hydrolase | 16 |
| Nucleic acid-binding, OB-fold | 13 |
| Histone-fold | 11 |
| Homeobox-like domain superfamily | 11 |
| Nucleotide-binding alpha-beta plait domain superfamily | 11 |
| RNA-binding domain superfamily | 11 |
| Helicase, C-terminal | 9 |
| RNA recognition motif domain | 9 |
| DEAD/DEAH box helicase domain | 8 |
| Helicase superfamily 1/2, ATP-binding domain | 8 |
| RNA helicase, DEAD-box type, Q motif | 7 |
| SANT/Myb domain | 7 |
| Ankyrin repeat | 6 |
| Armadillo-type fold | 6 |
| LSM domain superfamily | 6 |
| S-adenosyl-L-methionine-dependent methyltransferase | 6 |
| Zinc finger, RING/FYVE/PHD-type | 5 |
| ATP-dependent RNA helicase DEAD-box, conserved site | 4 |
| K Homology domain, type 1 superfamily | 4 |
| Poly(ADP-ribose) polymerase, catalytic domain | 4 |
| Poly(ADP-ribose) polymerase, regulatory domain superfamily | 4 |
| Tetratricopeptide-like helical domain superfamily | 4 |
| WGR domain superfamily | 4 |
| Winged helix-like DNA-binding domain superfamily | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, M.W.; Beh, L.Y.; Yerlici, V.T.; Fang, W.; Kulej, K.; Garcia, B.A.; Landweber, L.F. Exploration of the Nuclear Proteomes in the Ciliate Oxytricha trifallax. Microorganisms 2023, 11, 343. https://doi.org/10.3390/microorganisms11020343
Lu MW, Beh LY, Yerlici VT, Fang W, Kulej K, Garcia BA, Landweber LF. Exploration of the Nuclear Proteomes in the Ciliate Oxytricha trifallax. Microorganisms. 2023; 11(2):343. https://doi.org/10.3390/microorganisms11020343
Chicago/Turabian StyleLu, Michael W., Leslie Y. Beh, V. Talya Yerlici, Wenwen Fang, Katarzyna Kulej, Benjamin A. Garcia, and Laura F. Landweber. 2023. "Exploration of the Nuclear Proteomes in the Ciliate Oxytricha trifallax" Microorganisms 11, no. 2: 343. https://doi.org/10.3390/microorganisms11020343
APA StyleLu, M. W., Beh, L. Y., Yerlici, V. T., Fang, W., Kulej, K., Garcia, B. A., & Landweber, L. F. (2023). Exploration of the Nuclear Proteomes in the Ciliate Oxytricha trifallax. Microorganisms, 11(2), 343. https://doi.org/10.3390/microorganisms11020343