

Identification and Construction of Strong Promoters in Yarrowia lipolytica Suitable for Glycerol-Based Bioprocesses

 , , and
, , and 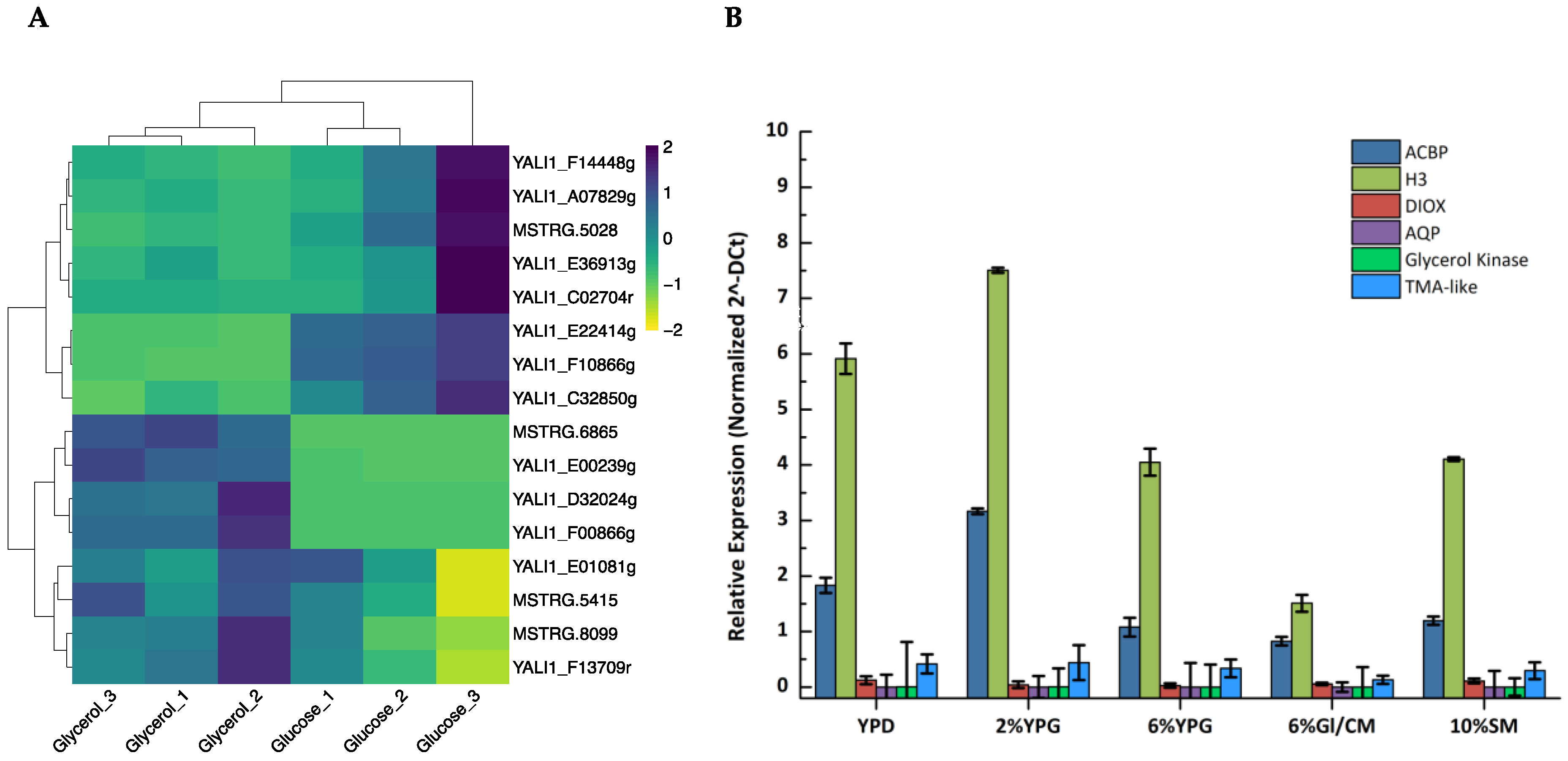{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
2.2. General Cloning
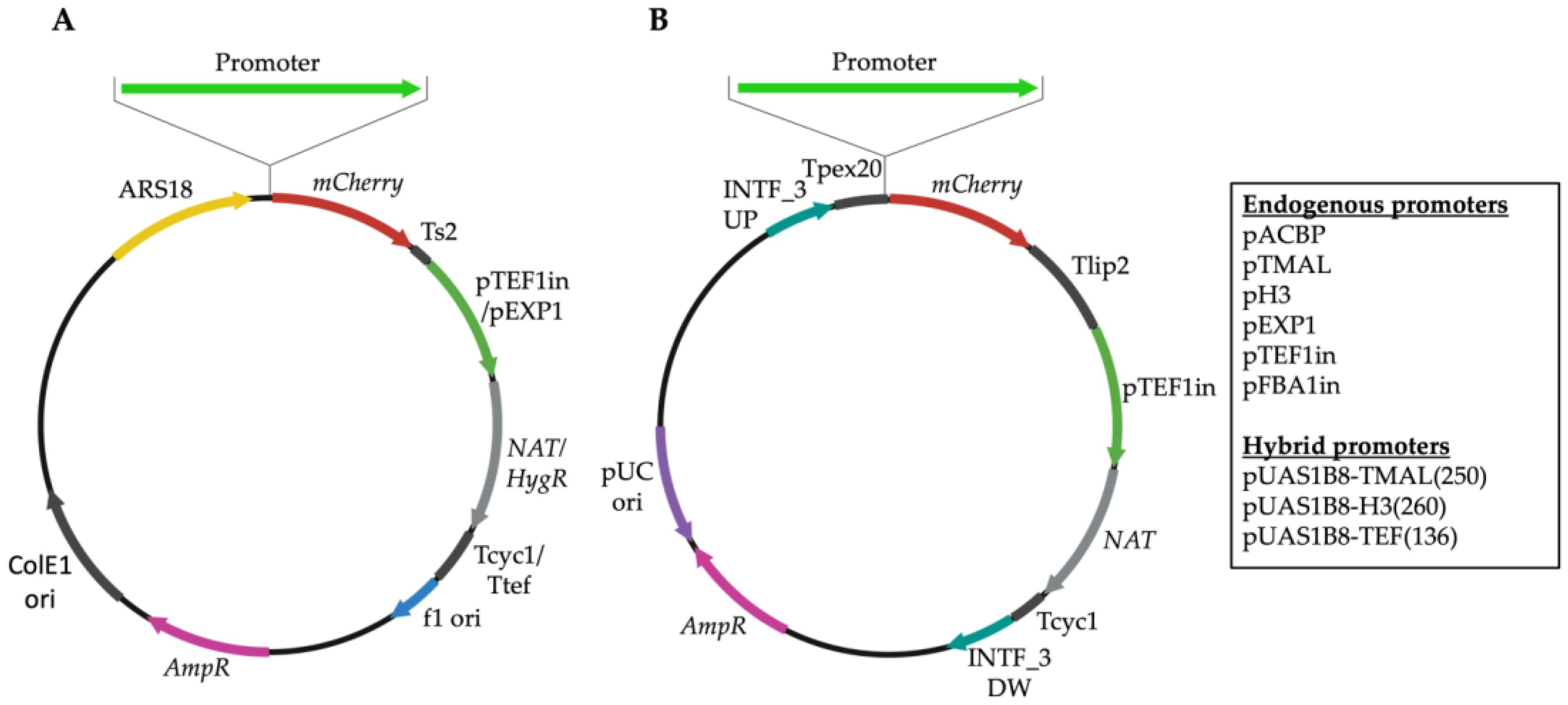
2.3. Construction of Yarrowia lipolytica Expression Vectors
2.4. Construction of Hybrid Promoters
2.5. Construction of Integration Vectors
2.6. Reverse Transcription-Quantitative PCR
2.7. Bioinformatic Analysis of Public Transcriptome Data
2.8. Flow Cytometry and Fluorescence Microscopy
2.9. Determination of Lipase Activity
3. Results
3.1. Identification of Highly Expressed Genes in Y. lipolytica Cultured in Glycerol Media
3.2. Development of Novel Promoter–Reporter Constructs
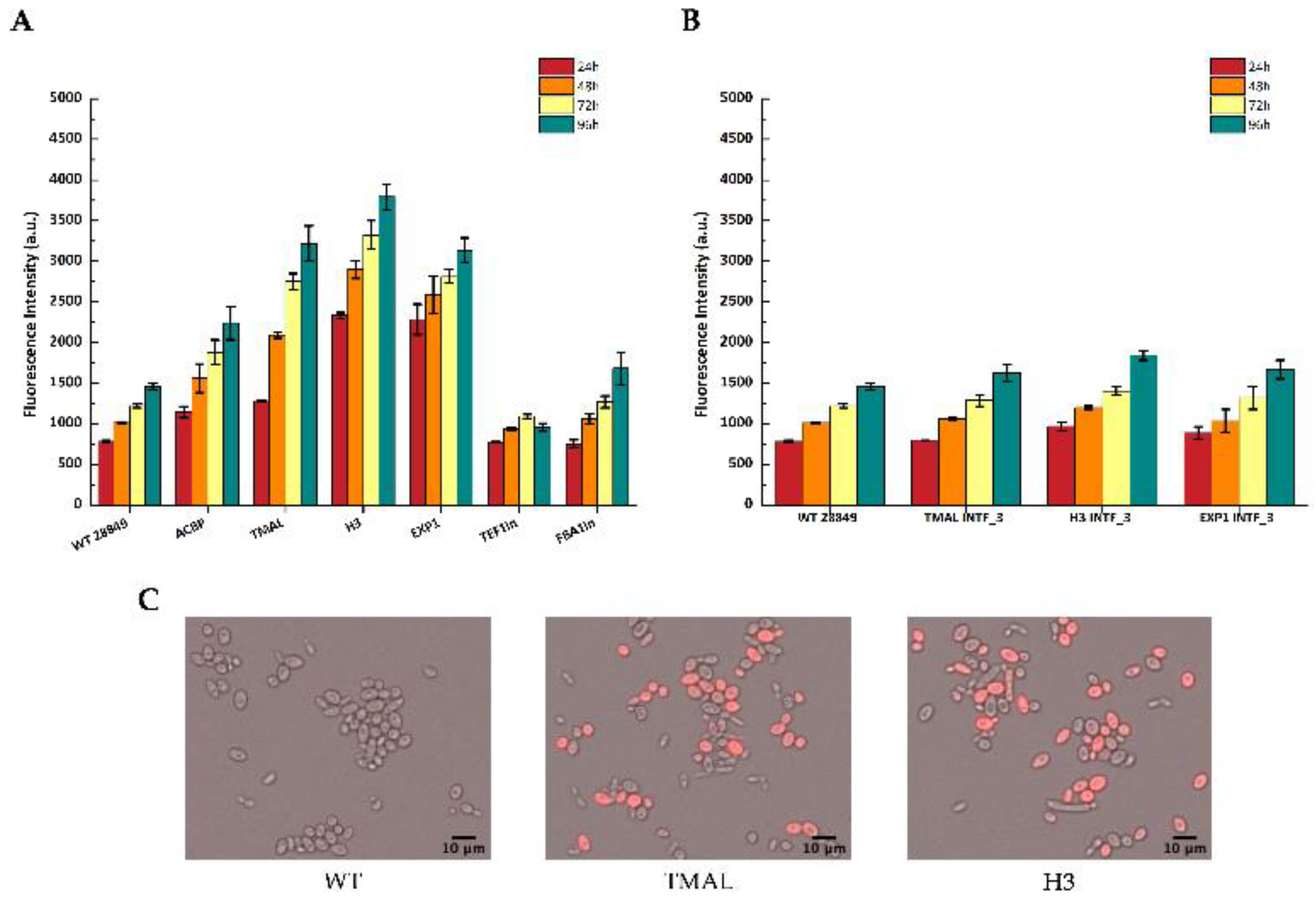
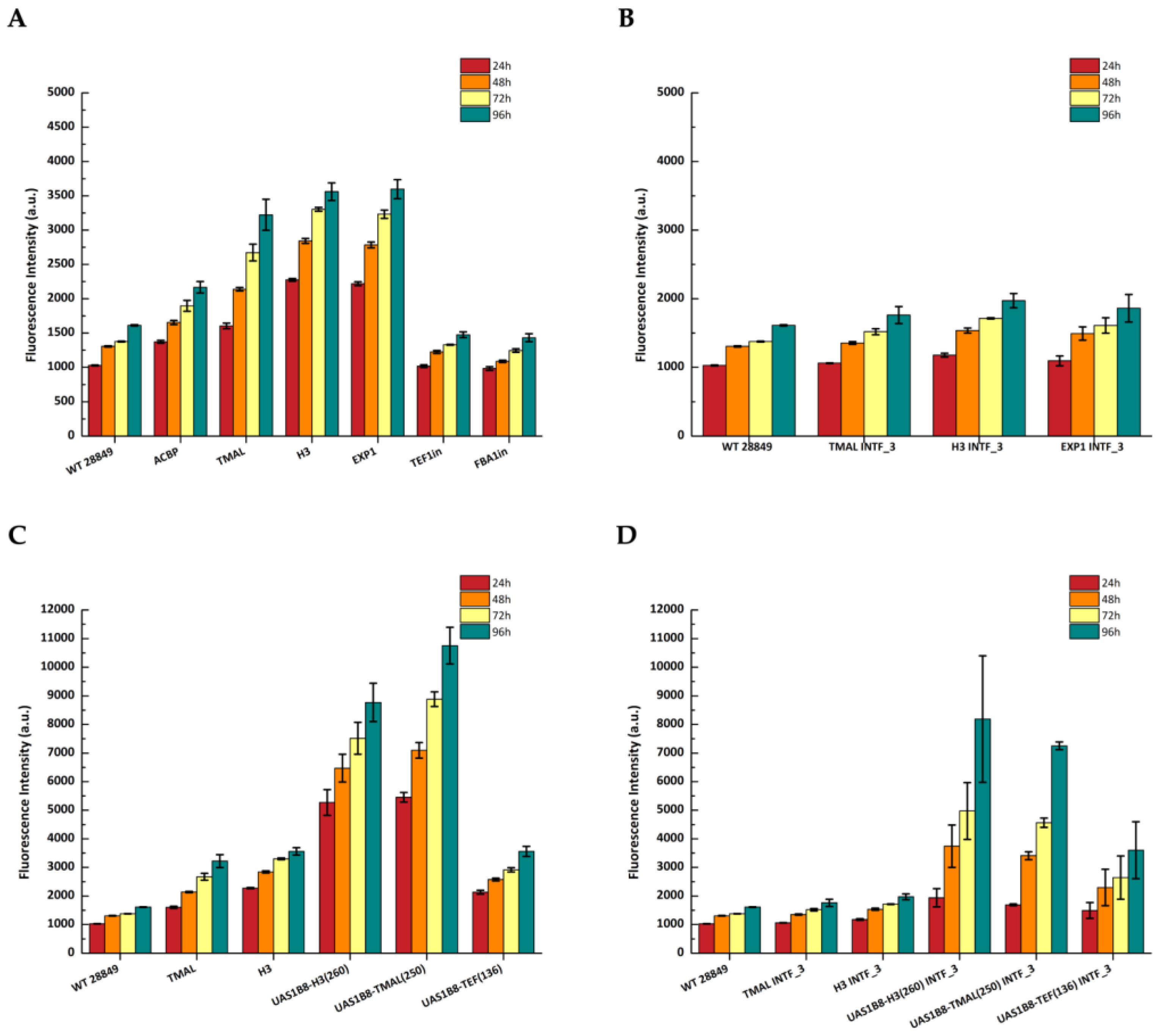
3.3. Reporter Gene Expression Driven by Selected Promoters
3.4. Development of Hybrid Promoters
3.5. Effect of Carbon Source on the Promoter Strength
3.6. Effect of Synthetic Medium on Promoter Strength
3.7. Expression of LIP2 from Integrated Expression Constructs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IEA. Renewables 2022. 2022. Available online: https://www.iea.org/reports/renewables-2022 (accessed on 16 January 2023).
- Demirbas, A.; Bafail, A.; Ahmad, W.; Sheikh, M. Biodiesel production from non-edible plant oils. Energy Explor. Exploit. 2016, 34, 290–318. [Google Scholar] [CrossRef] [Green Version]
- Machado Junior, F.R.S.; Michelon, M.; Dalcanton, F.; Furlong, E.B.; Burkert, J.F.M.; Burkert, C.A.V. Biomass production by Yarrowia lipolytica as a source of lipids: Bench scale cultivation on raw glycerol-based medium. Int. Food Res. J. 2015, 22, 1253–1260. [Google Scholar]
- Meher, L.C.; Vidya Sagar, D.; Naik, S.N. Technical aspects of biodiesel production by transesterification—A review. Renew. Sustain. Energy Rev. 2006, 10, 248–268. [Google Scholar] [CrossRef]
- Willke, T.; Vorlop, K.-D. Industrial bioconversion of renewable resources as an alternative to conventional chemistry. Appl. Microbiol. Biotechnol. 2004, 66, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mawgoud, A.M.; Markham, K.A.; Palmer, C.M.; Liu, N.; Stephanopoulos, G.; Alper, H.S. Metabolic engineering in the host Yarrowia lipolytica. Metab. Eng. 2018, 50, 192–208. [Google Scholar] [CrossRef]
- Beopoulos, A.; Cescut, J.; Haddouche, R.; Uribelarrea, J.-L.; Molina-Jouve, C.; Nicaud, J.-M. Yarrowia lipolytica as a model for bio-oil production. Prog. Lipid Res. 2009, 48, 375–387. [Google Scholar] [CrossRef]
- Celińska, E.; Grajek, W. A novel multigene expression construct for modification of glycerol metabolism in Yarrowia lipolytica. Microb. Cell Factories 2013, 12, 102. [Google Scholar] [CrossRef] [Green Version]
- Groenewald, M.; Boekhout, T.; Neuvéglise, C.; Gaillardin, C.; van Dijck, P.W.M.; Wyss, M. Yarrowia lipolytica: Safety assessment of an oleaginous yeast with a great industrial potential. Crit. Rev. Microbiol. 2014, 40, 187–206. [Google Scholar] [CrossRef]
- Hatti-Kaul, R.; Törnvall, U.; Gustafsson, L.; Börjesson, P. Industrial biotechnology for the production of bio-based chemicals—A cradle-to-grave perspective. Trends Biotechnol. 2007, 25, 119–124. [Google Scholar] [CrossRef]
- Levinson, W.E.; Kurtzman, C.P.; Kuo, T.M. Characterization of Yarrowia lipolytica and related species for citric acid production from glycerol. Enzym. Microb. Technol. 2007, 41, 292–295. [Google Scholar] [CrossRef]
- Madzak, C. Engineering Yarrowia lipolytica for Use in Biotechnological Applications: A Review of Major Achievements and Recent Innovations. Mol. Biotechnol. 2018, 60, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Workman, M.; Holt, P.; Thykaer, J. Comparing cellular performance of Yarrowia lipolytica during growth on glucose and glycerol in submerged cultivations. AMB Express 2013, 3, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodosiou, E. Engineering Strategies for Efficient Bioconversion of Glycerol to Value-Added Products by Yarrowia lipolytica. Catalysts 2023, 13, 657. [Google Scholar] [CrossRef]
- Choi, K.R.; Jang, W.D.; Yang, D.; Cho, J.S.; Park, D.; Lee, S.Y. Systems Metabolic Engineering Strategies: Integrating Systems and Synthetic Biology with Metabolic Engineering. Trends Biotechnol. 2019, 37, 817–837. [Google Scholar] [CrossRef] [PubMed]
- Markham, K.A.; Alper, H.S. Synthetic Biology Expands the Industrial Potential of Yarrowia lipolytica. Trends Biotechnol. 2018, 36, 1085–1095. [Google Scholar] [CrossRef]
- Tsirigka, A.; Theodosiou, E.; Patsios, S.I.; Tsoureki, A.; Andreadelli, A.; Papa, E.; Aggeli, A.; Karabelas, A.J.; Makris, A.M. Novel evolved Yarrowia lipolytica strains for enhanced growth and lipid content under high concentrations of crude glycerol. Microb. Cell Factories 2023, 22, 62. [Google Scholar] [CrossRef]
- Ledesma-Amaro, R.; Dulermo, T.; Nicaud, J.M. Engineering Yarrowia lipolytica to produce biodiesel from raw starch. Biotechnol. Biofuels 2015, 8, 148. [Google Scholar] [CrossRef] [Green Version]
- Papanikolaou, S.; Aggelis, G. Yarrowia lipolytica: A model microorganism used for the production of tailor-made lipids. Eur. J. Lipid Sci. Technol. 2010, 112, 639–654. [Google Scholar] [CrossRef]
- Yuzbasheva, E.Y.; Yuzbashev, T.V.; Perkovskaya, N.I.; Mostova, E.B.; Vybornaya, T.V.; Sukhozhenko, A.V.; Toropygin, I.Y.; Sineoky, S.P. Cell Surface Display of Yarrowia lipolytica Lipase Lip2p Using the Cell Wall Protein YlPir1p, Its Characterization, and Application as a Whole-Cell Biocatalyst. Appl. Biochem. Biotechnol. 2015, 175, 3888–3900. [Google Scholar] [CrossRef]
- Andriukonis, E.; Celiesiute-Germaniene, R.; Ramanavicius, S.; Viter, R.; Ramanavicius, A. From Microorganism-Based Amperometric Biosensors towards Microbial Fuel Cells. Sensors 2021, 21, 2442. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Holkenbrink, C.; Dam, M.I.; Kildegaard, K.R.; Beder, J.; Dahlin, J.; Doménech Belda, D.; Borodina, I. EasyCloneYALI: CRISPR/Cas9-Based Synthetic Toolbox for Engineering of the Yeast Yarrowia lipolytica. Biotechnol. J. 2018, 13, 1700543. [Google Scholar] [CrossRef] [Green Version]
- Larroude, M.; Trabelsi, H.; Nicaud, J.-M.; Rossignol, T. A set of Yarrowia lipolytica CRISPR/Cas9 vectors for exploiting wild-type strain diversity. Biotechnol. Lett. 2020, 42, 773–785. [Google Scholar] [CrossRef] [Green Version]
- Madzak, C.; Blanchin-Roland, S.; Cordero Otero, R.R.; Gaillardin, C. Functional analysis of upstream regulating regions from the Yarrowia lipolytica XPR2 promoter. Microbiology 1999, 145 Pt 1, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Blazeck, J.; Liu, L.; Redden, H.; Alper, H. Tuning gene expression in Yarrowia lipolytica by a hybrid promoter approach. Appl. Environ. Microbiol. 2011, 77, 7905–7914. [Google Scholar] [CrossRef] [Green Version]
- Darvishi, F.; Ariana, M.; Marella, E.R.; Borodina, I. Advances in synthetic biology of oleaginous yeast Yarrowia lipolytica for producing non-native chemicals. Appl. Microbiol. Biotechnol. 2018, 102, 5925–5938. [Google Scholar] [CrossRef]
- Hong, S.P.; Seip, J.; Walters-Pollak, D.; Rupert, R.; Jackson, R.; Xue, Z.; Zhu, Q. Engineering Yarrowia lipolytica to express secretory invertase with strong FBA1IN promoter. Yeast 2012, 29, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Tai, M.; Stephanopoulos, G. Engineering the push and pull of lipid biosynthesis in oleaginous yeast Yarrowia lipolytica for biofuel production. Metab. Eng. 2013, 15, 1–9. [Google Scholar] [CrossRef]
- Le Hir, H.; Nott, A.; Moore, M.J. How introns influence and enhance eukaryotic gene expression. Trends Biochem. Sci. 2003, 28, 215–220. [Google Scholar] [CrossRef]
- Blazeck, J.; Reed, B.; Garg, R.; Gerstner, R.; Pan, A.; Agarwala, V.; Alper, H.S. Generalizing a hybrid synthetic promoter approach in Yarrowia lipolytica. Appl. Microbiol. Biotechnol. 2013, 97, 3037–3052. [Google Scholar] [CrossRef]
- Shabbir Hussain, M.; Gambill, L.; Smith, S.; Blenner, M.A. Engineering Promoter Architecture in Oleaginous Yeast Yarrowia lipolytica. ACS Synth. Biol. 2016, 5, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.C.; Yang, B.C.; Kuo, T.T. One-step transformation of yeast in stationary phase. Curr. Genet. 1992, 21, 83–84. [Google Scholar] [CrossRef]
- Jost, B.; Holz, M.; Aurich, A.; Barth, G.; Bley, T.; Müller, R.A. The influence of oxygen limitation for the production of succinic acid with recombinant strains of Yarrowia lipolytica. Appl. Microbiol. Biotechnol. 2015, 99, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Fournier, P.; Guyaneux, L.; Chasles, M.; Gaillardin, C. Scarcity of ars sequences isolated in a morphogenesis mutant of the yeast Yarrowia lipolytica. Yeast 1991, 7, 25–36. [Google Scholar] [CrossRef]
- Curran, K.A.; Morse, N.J.; Markham, K.A.; Wagman, A.M.; Gupta, A.; Alper, H.S. Short Synthetic Terminators for Improved Heterologous Gene Expression in Yeast. ACS Synth. Biol. 2015, 4, 824–832. [Google Scholar] [CrossRef]
- Borkowska, M.; Białas, W.; Celińska, E. A new set of reference genes for comparative gene expression analyses in Yarrowia lipolytica. FEMS Yeast Res. 2020, 20, foaa059. [Google Scholar] [CrossRef]
- Felix Krueger, F.J.; Ewels, P.; Afyounian, E.; Schuster-Boeckler, B. FelixKrueger/TrimGalore, Version 0.6.7; DOI via Zenodo (0.6.7). 2021. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 20 February 2020).
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-Level Expression Analysis of RNA-Seq Experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. 2022. Available online: https://www.R-Project.Org/ (accessed on 12 April 2023).
- Wickham, H. ggplot2; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar] [CrossRef]
- Kolde, R. pheatmap: Pretty Heatmaps. R package, version 1.0. 12; R-project. org/package pheatmap; CRAN. 2019.
- Duquesne, S.; Bordes, F.; Fudalej, F.; Nicaud, J.-M.; Marty, A. The yeast Yarrowia lipolytica as a generic tool for molecular evolution of enzymes. Methods Mol. Biol. 2012, 861, 301–312. [Google Scholar] [PubMed] [Green Version]
- Vernis, L.; Abbas, A.; Chasles, M.; Gaillardin, C.M.; Brun, C.; Huberman, J.A.; Fournier, P. An origin of replication and a centromere are both needed to establish a replicative plasmid in the yeast Yarrowia lipolytica. Mol. Cell. Biol. 1997, 17, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Darvishi, F.; Nahvi, I.; Zarkesh-Esfahani, H.; Momenbeik, F. Effect of Plant Oils upon Lipase and Citric Acid Production in Yarrowia lipolytica Yeast. J. Biomed. Biotechnol. 2009, 2009, 562943. [Google Scholar] [CrossRef]
- Shabbir Hussain, M.; Wheeldon, I.; Blenner, M.A. A Strong Hybrid Fatty Acid Inducible Transcriptional Sensor Built From Yarrowia lipolytica Upstream Activating and Regulatory Sequences. Biotechnol. J. 2017, 12, 1700248. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.C.; Viana, R.; Palma, M.; Oliveira, J.; Galocha, M.; Mota, M.N.; Couceiro, D.; Pereira, M.G.; Antunes, M.; Costa, I.V.; et al. YEASTRACT+: A portal for the exploitation of global transcription regulation and metabolic model data in yeast biotechnology and pathogenesis. Nucleic Acids Res. 2023, 51, D785–D791. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.M.; Hussain, M.S.; Blenner, M.; Wheeldon, I. Synthetic RNA Polymerase III Promoters Facilitate High-Efficiency CRISPR-Cas9-Mediated Genome Editing in Yarrowia lipolytica. ACS Synth. Biol. 2016, 5, 356–359. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgiadis, I.; Tsiligkaki, C.; Patavou, V.; Orfanidou, M.; Tsoureki, A.; Andreadelli, A.; Theodosiou, E.; Makris, A.M. Identification and Construction of Strong Promoters in Yarrowia lipolytica Suitable for Glycerol-Based Bioprocesses. Microorganisms 2023, 11, 1152. https://doi.org/10.3390/microorganisms11051152
Georgiadis I, Tsiligkaki C, Patavou V, Orfanidou M, Tsoureki A, Andreadelli A, Theodosiou E, Makris AM. Identification and Construction of Strong Promoters in Yarrowia lipolytica Suitable for Glycerol-Based Bioprocesses. Microorganisms. 2023; 11(5):1152. https://doi.org/10.3390/microorganisms11051152
Chicago/Turabian StyleGeorgiadis, Ioannis, Christina Tsiligkaki, Victoria Patavou, Maria Orfanidou, Antiopi Tsoureki, Aggeliki Andreadelli, Eleni Theodosiou, and Antonios M. Makris. 2023. "Identification and Construction of Strong Promoters in Yarrowia lipolytica Suitable for Glycerol-Based Bioprocesses" Microorganisms 11, no. 5: 1152. https://doi.org/10.3390/microorganisms11051152
APA StyleGeorgiadis, I., Tsiligkaki, C., Patavou, V., Orfanidou, M., Tsoureki, A., Andreadelli, A., Theodosiou, E., & Makris, A. M. (2023). Identification and Construction of Strong Promoters in Yarrowia lipolytica Suitable for Glycerol-Based Bioprocesses. Microorganisms, 11(5), 1152. https://doi.org/10.3390/microorganisms11051152