1. Introduction
The progressive loss of muscle strength and functionality that characterizes different physiological (aging) and pathological (sarcopenia, cachexia, idiopathic chronic fatigue, myasthenia gravis) statuses is thought to be due to alterations in mitochondria, including reduced mitochondrial content, impaired oxidative capacity, and increased oxidative damage [
1,
2,
3,
4,
5].
As a deficit of vitamin D is often associated with these muscle-affecting conditions, vitamin D supplementation has been proposed as a potential treatment to improve muscle strength, albeit with conflicting results. In particular, supplementation of vitamin D appears to be efficacious in restoring muscle function in elderly but not in cachectic muscle [
6,
7,
8,
9,
10,
11,
12]. Several works have investigated the effects of vitamin D on mitochondrial function in muscle-derived cells. In particular, the treatment of both human primary and C2C12 myoblasts with 1α,25-dihydroxyvitamin D3 (1,25VD) increases mitochondrial function [
13,
14,
15]. This activity of 1,25VD on mitochondria is even effective in mitigating the dysfunctional metabolism induced by the treatment of myoblasts with conditioned medium derived from cancer cells [
16]. Additionally, 1,25VD increases mitochondrial respiration in myotubes derived from the differentiation of both human and C2C12 myoblasts [
14,
17]. Notably, in both C2C12 myoblasts and myotubes and human myoblasts, the silencing of the vitamin D receptor (VDR) results in a significant reduction in mitochondrial respiration [
13,
15]. Accordingly, the reduction in mitochondrial respiration in human myoblasts upon treatment with cholecalciferol (VD3) or 25-hydroxyvitamin D3 (25VD) led to the hypothesis that the increase in mitochondrial oxygen consumption rate (OCR) depends on the high binding affinity of 1,25VD for VDR relative to the lower affinities of other metabolites [
13].
However, despite the evidence correlating vitamin D deficiency with muscular impairment and the undisputed importance of the VD/VDR system for skeletal muscle health, treatment of C2C12 myotubes with 1,25VD negatively impinges on myotube size [
11,
18]. In contrast, the upstream metabolites of 1,25VD, namely VD3 and 25VD, have an anti-atrophic activity on the same in vitro cellular model [
18,
19].
Since mitochondrial dysfunction can induce muscle atrophy in several conditions [
20], in the present study, we investigated the effects of 1,25VD and its upstream metabolites VD3 and 25VD on mitochondrial function and organization in C2C12 myotubes.
2. Materials and Methods
2.1. Reagents
VD3, 25VD, and 1,25VD were purchased from Merck Life Sciences (Milan, Italy) and dissolved in ethanol. Protein kinase C (PKC) inhibitor (GÖ6850) and c-Jun N-terminal kinase (JNK) inhibitor (SP600125) were from Cayman Chemical (Ann Arbor, MI, USA). N-Acetyl-L-Cysteine (NAC), MitoTracker Red CMXRos, and CellROX R Deep Red Reagent were from Invitrogen (Thermo Fisher Scientific, Waltham, MA, USA). JC-1 was from Adipogen Life Sciences (Liestal, Switzerland). Anti-LC3B antibody was from Proteintech (Rosemont, IL, USA), antitubulin antibody was from Santa Cruz Biotechnology (Dallas, TX, USA), and anti-VDAC and antiphospho-(Ser) PKC substrates antibodies were from Cell Signaling Technology (Danvers, MA, USA). MiR05 was purchased from Oroboros Instruments (Innsbruck, Austria). All other reagents, unless otherwise stated, were from Merck Life Sciences (Milan, Italy).
2.2. Cell Culture and Myotube Analysis
C2C12 myoblasts (ECACC, Porton Down, Salisbury, UK) were grown at low density in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, Thermo Fisher Scientific, Waltham, MA, USA), 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL antimycotic in a humidified 5% CO
2 incubator at 37 °C. To induce differentiation, cells were allowed to become confluent, and the medium was switched to differentiation medium (DM), consisting of DMEM supplemented with 2% horse serum (GE Healthcare BioSciences, Uppsala, Sweden), penicillin, streptomycin, and antimycotic as described above. Unless otherwise specified, myotubes were in treated serum-free medium after at least five days of differentiation. Control cells were treated with 0.1% ethanol. Myotube diameters were measured with JMicroVision software (v. 1.3.4, University of Geneva, Switzerland) as previously described [
21]. For every experiment assessing myotube diameters, at least 10 myotubes for each field, five different fields for each replicate, and three technical replicates for each treatment were measured. Displayed data are the average of three independent experiments.
2.3. DNA Extraction and Analysis
DNA was extracted from myotubes using the NucleoSpin Tissue purification kit (Macherey-Nagel, Düren, Germany) according to the manufacturer’s instructions. The isolated DNA was eluted in 60 µL of elution buffer. To investigate changes in mitochondria number, real-time PCR was performed with the StepOnePlus Real-time PCR System (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA), using the following TaqMan probes (Thermo Fisher Scientific, Waltham, MA, USA): Rps18 (Mm00507222_s1) and Cox2 (Mm03294838_g1).
2.4. RNA Extraction and Analysis
Total RNA from myotubes was extracted by RNAzol (Merck Life Sciences, Milan, Italy). The RNA was retrotranscribed with high-capacity cDNA Reverse Transcription Kit (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA), and real-time PCR was performed with the StepOnePlus Real-time PCR System (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA), using the following TaqMan probes (Thermo Fisher Scientific, Waltham, MA, USA): Opa1 (Mm00453873_m1), Mff (Mm01273401_m1), Mfn2 (Mm00500120_m1), FbxO32 (Atrogin-1, Mm00499523_m1), Trim63 (MuRF-1, Mm01185221_m1), and Gusb (Mm01197698_m1).
2.5. Mitochondrial Membrane Potential Analysis
Mitochondrial membrane potential in C2C12 myotubes was assessed using the JC-1 mitochondrial membrane potential assay kit (Adipogen Life Sciences, Liestal, Switzerland), according to the manufacturer’s instructions. Briefly, 10 µg/mL JC-1 was added to the culture medium and gently mixed. Cells were then incubated in a CO2 incubator at 37 °C for 15 min. Images were acquired with a fluorescence microscope (EVOS™ XL Core Imaging System, Thermo Fisher Scientific, Waltham, MA, USA). Red emission of the dye represented a potential-dependent aggregation of JC-1 in the mitochondria. Conversely, green fluorescence appearing in the cytosol after mitochondrial membrane depolarization represented the monomeric form of JC-1. The average intensity of red and green fluorescence was measured using ImageJ software, and the ratio of JC-1 aggregate (red) to monomer (green) intensity was then calculated. For every experiment assessing mitochondrial membrane potential, at least five myotubes in each field, five different fields for each replicate, three technical replicates for each treatment were measured. The final data are the average of three independent experiments.
2.6. Determination of Reactive Oxygen Species (ROS) Production
To measure cellular ROS production, C2C12 myotubes were stained with CellROX R Deep Red Reagent (Thermo Fisher Scientific, Waltham, MA, USA) for 30 min at 37 °C and washed with PBS. Fluorescent images were taken using fluorescence microscope EVOS™ XL (Thermo Fisher Scientific, Waltham, MA, USA), and the mean fluorescence signal intensity was measured using ImageJ (v. 1.53j, National Institutes of Health, USA). For every experiment assessing cellular oxidative stress, at least five myotubes in each field, five different fields for each replicate, three technical replicates for each treatment were measured. The final data are the average of three independent experiments.
2.7. Mitochondrial Morphology and Mitophagy Evaluation
Fluorescent microscopy was performed to identify variations in both mitochondrial morphology and mitophagy. For mitochondrial network visualization, C2C12 myotubes were incubated with 100 nM MitoTracker Red CMXRos (Thermo Fisher Scientific, Waltham, MA, USA) for 45 min at 37 °C, 5% CO2. To analyze mitophagy, cells were washed in PBS, fixed with 4% paraformaldehyde for 10 min, and permeabilized for 5 min with 0.2% Triton X-100 (Merck Life Sciences, Milan, Italy). Blocking was then performed with 4% bovine serum albumin for 30 min. Subsequently, cells were incubated with a primary antibody directed against LC3B (microtubule-associated protein 1 light chain-3B) overnight at 4 °C and followed by the appropriate Alexa Fluor Dye-conjugated secondary antibody (488 antirabbit 1:400; Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at room temperature. Nuclei were counterstained with DAPI (1:100, Thermo Fisher Scientific, Waltham, MA, USA), and images were acquired with a Leica dm 5500b fluorescence microscope (Leica, Wetzlar, Germany) equipped with Leica Application Suite X software (v.3.7.4.23463, Leica, Wetzlar, Germany), using a 40× objective. For every experiment assessing mitochondrial morphology and mitophagy, at least two myotubes in each field, five different fields for each replicate, three technical replicates for each treatment were measured. The final data are the average of three independent experiments.
Mitochondrial morphology was analyzed using the Mitochondrial Network Analysis tool (MiNA, v.3.0.1), a macro tool developed for ImageJ, as previously described [
22]. Briefly, fluorescence images were processed removing the background and then skeletonized, and the resulting skeleton was analyzed using the Analyze Skeleton plugin included in the ImageJ software.
To quantify mitophagy, Pearson’s correlation coefficient of the fluorescent signals in both channels was calculated through the JACoP plugin [
23]. This coefficient describes the correlation between the intensity distribution or pattern overlap in two channels in terms of a least-squares fit. This value can be between −1 and 1, and
R = 1 indicates a complete correlation between the two channels. Finally, Pearson’ s coefficient indicates the overlap of the signals and represents the degree of colocalization.
2.8. Co-Immunoprecipitation Assay
For the co-immunoprecipitation assay, 15 min before the end of the indicated treatments, 1 mM of the chemical cross-linker 3-3′-dithiodipropionic acid di-(N-hydroxysuccinimide ester) was added. Cells were resuspended in ice-cold 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered saline (HBS: 150 mM NaCl, 20 mM HEPES) supplemented with protease inhibitor cocktail and lysed by passing through a 29-gauge needle 30 times. Nuclei and cell debris were spun down at 500× g for 5 min. Protein were quantified by BCA assay (ThermoFisher Scientific, Waltham, MA, USA), and 250 μg of proteins from each lysate were incubated for 4 h with primary anti-LC3B antibody (1:250) on a rotating wheel. To capture the immunocomplexes, 20 μL of Protein G sepharose Fast Flow were added to each sample and left under rotation overnight at 4 °C. After two washes with HBS, immunocomplexes were collected by centrifugation and eluted in 30 μL of sample buffer (2% sodium dodecyl sulfate, 150 mM dithiothreitol, and 0.01% bromophenol blue) at 95 °C for 10 min. Finally, the association LC3B-VDAC was evaluated by western blotting. First, 15 μg of the whole cell lysate (input) and all the immunoprecipitated fraction were separated by 15% SDS-PAGE and transferred to polyvinylidene difluoride filters (PVDF) (Hybond-P; GE Healthcare, Little Chalfont, Buckinghamshire, UK). Membranes were then saturated with 4% bovine serum albumin (BSA), incubated with anti-LC3B (1:600), anti-VDAC (1:1000), or antitubulin (1:1000) antibodies overnight, washed with Tris-buffered saline (TBS) 0.1% Tween, and incubated with the appropriate secondary antibody (1:3000; Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at room temperature. Specific signals were visualized with Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA), and acquired with ChemiDoc Touch (Bio-Rad, Hercules, CA, USA).
2.9. Western Blotting
At the end of the indicated treatments, cells were washed in ice-cold PBS and solubilized with a lysis buffer containing 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 160 mM NaCl, 20 mM Tris-HCl, pH 7.4, and supplemented with protease inhibitor cocktail. Lysates were stirred at 4° C for 15 min and centrifuged at 15,000× g for 15 min at 4° C. Protein concentration was determined by BCA protein assay kit. Proteins (20 μg protein/lane) were separated by 10% SDS-PAGE and transferred to PVDF. Membranes were saturated with 4% BSA, incubated with the antiphospho-(Ser) PKC substrates and antitubulin antibodies (1:1000) overnight, washed with TBS 0.1% Tween, incubated with the appropriate secondary antibody (1:3000) for 1 h at room temperature, visualized with Western Lightning Chemiluminescence Reagent Plus, acquired with ChemiDoc Touch, and analyzed with ImageLab (Bio-Rad, Hercules, CA, USA).
2.10. Intact Cell Respiration Using High-Resolution Respirometry
We determined cellular respiration using an Oroboros oxygraph-2K high-resolution respirometer (Oroboros Instruments, Innsbruck, Austria) and substrate, uncoupler, inhibitor, titration (SUIT) protocols [
24,
25,
26]. To assess mitochondrial respiration in intact cells, we used the SUIT-003_O2_ce_D012 protocol, as recommended by the manufacturer of the Oroboros instrument. At the end of the treatments, C2C12 myotubes were trypsinized, centrifuged at 300×
g for 5 min, resuspended in mitochondrial respiration medium MiR05 (0.5 mM EGTA, 3.0 mM MgCl
2·6H
2O, 60 mM potassium lactobionate, 20 mM taurine, 10 mM KH
2PO
4, 20 mM HEPES, 110 mM sucrose, 1 g/L bovine serum albumin, pH 7.1) and transferred to the chambers of the Oroboros oxygraph. Control and treated samples were assessed simultaneously. After initial stabilization of O
2 flux, pyruvate (5 mM) was used to sustain TCA-linked respiration in MiR05 medium. ATP synthetase inhibitor, oligomycin (Omy), was added at 5 nM final concentration, and oxygen consumption was quantified to determine the oligomycin-sensitive and -insensitive respiration. Protonophore (H
+ ionophore) and uncoupler of oxidative phosphorylation, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (U) was then added at 0.5 μM increments to achieve maximum respiration to quantify maximum respiratory capacity. This was followed by rotenone (Rot) 500 nM final concentration, to inhibit complex I of the electron transport chain (ETC), and then 2.5 μM antimycin A (Ama), which inhibits complex III, was added to determine the nonmitochondrial respiration (ROX). Oxygen consumption rates were calculated using accompanying software (DatLab7, Oroboros, Innsbruck, Austria) as described earlier [
24]. Rates of O
2 consumption (flux) were normalized to total protein content. Briefly, at the end of the experimental procedure, the cellular suspension from the two chambers was centrifuged at 1000×
g for 5 min. The cellular pellet was lysed in 200 µL of lysis buffer (10 mM HEPES, 60 mM KCl, 1 mM EDTA, 0.075% NP40, 1 mM DTT) and then centrifuged at 15,000×
g for 15 min at 4 °C. The concentration of the protein in the supernatant was measured with Bradford Reagent (Merck Life Sciences, Milan, Italy).
2.11. Mitochondrial Oxidative Phosphorylation (OXPHOS) in Permeabilized Myotubes
To quantify mitochondrial function in permeabilized myotubes, we performed high-resolution respirometry using the SUIT-008_O2_ce-pce_D025 protocol designed by Oroboros Instruments to standardize the evaluation of OXPHOS and electron transfer capacities linked to the complex I and II. Briefly, C2C12 myotubes were trypsinized, centrifuged at 300×
g for 5 min, and resuspended in mitochondrial respiration medium MiR05. After the transfer in the oxygraph, cell permeabilization by 10 µg/mL of digitonin was followed by the addition of mitochondrial substrates malate (2 mM) and pyruvate (5 mM) to generate NADH, substrate for complex I. To maintain the oxidation of pyruvate to acetyl CoA via pyruvate dehydrogenase, malate was added simultaneously as a source of oxaloacetate to metabolize acetyl CoA to citrate. Leakage respiration was measured in the presence of pyruvate and malate without ADP. The addition of ADP at a saturating concentration (2.5 mM) was used to quantify OXPHOS capacity, that is, the capacity of oxidation (electron transport down the gradient along the ETC) and ATP synthesis of complex I. The integrity of the mitochondrial membrane was tested by adding 10 µM cytochrome c. Malate, pyruvate, and glutamate (10 mM), provided as complex I substrates, were followed by succinate (10 mM) as a complex II substrate to quantify the OXPHOS capacity of both complexes I and II. The contribution of complex II to the OXPHOS was obtained as the difference of oxygen flux before and after the addition of succinate. Oxidation and phosphorylation were uncoupled with 0.5 µM FCCP to measure maximal respiration. The uncoupled rate of oxygen consumption of complex I (the rotenone-sensitive rate) was determined by the absolute decrease from the maximum uncoupling rate of all substrates. The rotenone-insensitive rate is the uncoupled rate of oxygen consumption of complex II. After the injection of antimycin, the uncoupled oxidation rate of complex IV was calculated by subtracting the azide-insensitive rate from the tetramethyl phenylene diamine (TMPD, 0.5 mM) + ascorbate (2 mM) rate. Oxygen consumption rates were calculated using accompanying software (DatLab7, Oroboros, Innsbruck, Austria) as described earlier [
24] and normalized to protein total content.
2.12. Mitochondrial ROS Production
Hydrogen peroxide (H2O2) production was measured in the presence of horseradish peroxidase (HRP) and Amplex Ultrared Reagent (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA), which react in a 1:1 stoichiometry with H2O2 to produce highly fluorescent resorufin. H2O2 production rates were measured in the oxygraphic chamber, multiplexed with measurements of oxygen consumption using the SUIT-006_AmR_mt_D048 protocol, as recommended by Oroboros Instruments (Innsbruck, Austria). Superoxide dismutase (5 U/mL), horseradish peroxidase (1 U/mL), and Amplex Ultra Red (10 µM) were added into the oxygraphic chambers containing C2C12 myotubes. After permeabilization with digitonin (10 µg/mL), pyruvate (5 mM) and malate (2 mM) were added to evaluate the leakage state (LEAK), while the following supplementation of ADP (2.5 mM) induced the oxygen and H2O2 flux relative to the maximum OXPHOS capacity. The LEAK state dependent on the ATP-synthase inhibition was obtained with the addition of 5 µM oligomycin. The fluorescent signal was adjusted for background auto-oxidation and calibrated to a standard curve. Rates of H2O2 production (flux) were normalized to protein total content.
2.13. Statistical Analysis
The investigators quantifying the experimental outcomes were blind to the treatments, and the statistic evaluation of the experimental data was performed by another investigator not directly involved in data collection and parameters measurement.
Data are presented as the mean ± SEM. Outliers in the measurements were identified by mean of the interquartile range (IQR), as either below Q1 − 1.5 IQR or above Q3 + 1.5 IQR and excluded from the analysis. The variation among groups was evaluated using Student’s t test or one-way ANOVA test followed by Tukey’s multiple comparisons test, as appropriate. Statistical significance was assumed for p < 0.05. All statistical analyses were performed with GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA).
4. Discussion
Vitamin D, especially in the form of 1,25VD, is often regarded as being endowed with antioxidant capacity which allows it to mitigate ROS production and prevent oxidative stress in skeletal muscle [
31,
32]. However, in this work, we demonstrated that 1,25VD induces production of ROS in C2C12 myotubes that, in turn, cause the atrophic phenotype in these cells.
The notion that mitochondrial dysfunction can lead to muscle wasting [
33] applied to our model of 1,25VD-induced atrophy [
18], prompted us to assess if 1,25VD impinged mitochondrial function in C2C12 myotubes. To our surprise, besides a strong depolarization of mitochondrial membranes (
Figure 1), 1,25VD failed to elicit any significant effect on myotube mitochondria in intact cells (
Figure 2 and
Figure 4). Only in permeabilized cells, i.e., a system that resembles the analysis of isolated mitochondria, we did observe a decrease in the maximal oxidative phosphorylation capacity of complex I (
Figure 3c). Therefore, the drop in mitochondrial membrane potential could hardly be a direct consequence of mitochondrial damage. Since it has been suggested that ROS production is upstream of mitochondrial depolarization [
34], we assessed the effect of 1,25VD on ROS production. In line with the limited effects on mitochondrial respiration, 1,25VD did not induce mitochondria-generated ROS (
Figure 6c,d). Nevertheless, the intensity of the fluorescence of the probe measuring ROS production dramatically increased after 1,25VD treatment (
Figure 6a,b), and the use of a generic antioxidant such as NAC completely prevented the atrophy associated with 1,25VD treatment (
Figure 7a–f), suggesting that 1,25VD induces the generation of ROS from cellular sources other than mitochondria. Consistently, the inhibition of mitochondrial ROS was not equally effective in protecting C2C12 myotubes from 1,25VD-induced atrophy (
Figure 7g–k). Despite the fact that mitochondria are often considered the most relevant ROS source, in skeletal muscle cells, NAD(P)H oxidases (NOXs) have recently emerged as an equally important source of ROS able to crosstalk with mitochondria to exacerbate ROS production [
27]. Although the specific role and activation pathways of each NOX isoform in skeletal muscle have been only partially elucidated, one possible mechanism of activation of NOX2 is mediated by activation of classical PKCs [
28]. The involvement of PKC in 1,25VD-mediated MAP kinase activation in chicken myoblasts [
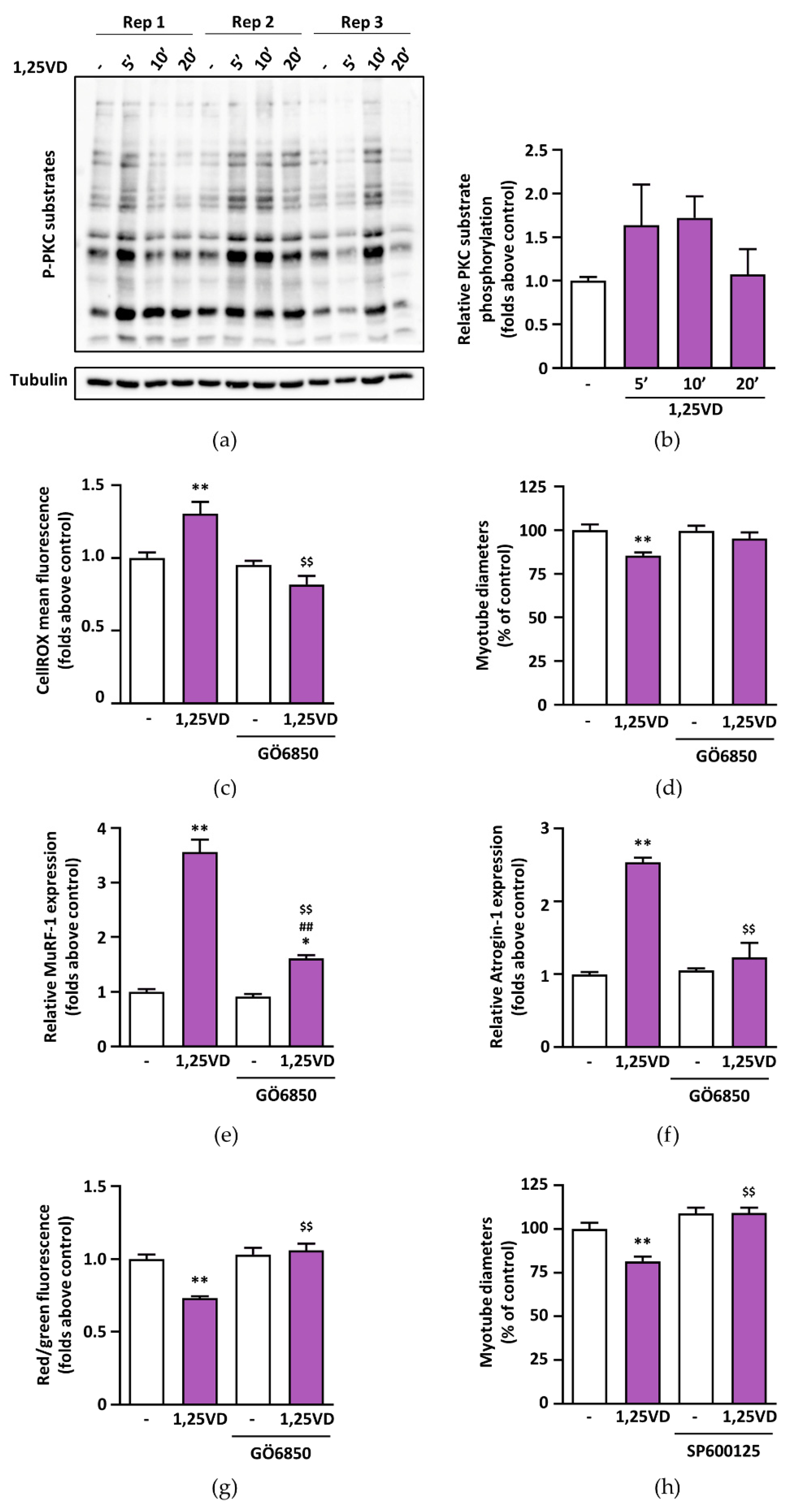
35] and the tendency of 1,25VD to phosphorylate PKC substrates in C2C12 myotubes (
Figure 8a,b) led us to speculate that 1,25VD-induced ROS production depends on PKC activation. In turn, ROS can lead to muscle atrophy via activation of JNK [
29,
30]. The hypothesis that 1,25VD-induced atrophy could depend on the activation of a PKC-ROS-JNK pathway is supported by the rescue of the atrophic phenotype when myotubes were treated with specific PKC and JNK inhibitors (
Figure 8c–h).
We previously demonstrated that 1,25VD-induced atrophy was accompanied by a block of the autophagic flux in C2C12 myotubes [
18]. Here, we observed that the blocking of the autophagic flux involves mitochondria as well (
Figure 5). We speculate that 1,25VD-induced mitochondria depolarization is a trigger for mitophagy [
36]. However, whether 1,25VD-induced block of mitophagy is under direct control of ROS is still to be determined. Altogether, these data deepen our knowledge of the mechanisms underlying 1,25VD-induced atrophy in C2C12 myoblasts.
The contrasting conclusions of our work compared with those of previously published studies probably depend, in large part, on differences in the methodologies or cells used. Indeed, the majority of the works reporting positive activity of 1,25VD on mitochondria were performed on undifferentiated myoblasts [
13,
16], while our data, as well as previously published data demonstrating the atrophic activity of 1,25VD [
18], were obtained on differentiated myotubes, a cellular model which is closer than myoblasts to adult muscle fibers [
19]. Indeed, the content, network shape, and function of mitochondria dramatically change with differentiation from myoblasts to myotubes [
37]. Accordingly, when analyzing the OCR in myoblasts treated with 1,25VD, we found a moderate increase in the maximal mitochondrial respiration (data not shown), in accordance with previously reported findings [
13]. In addition to the basal different content and activity of mitochondria in myoblasts vs. myotubes, we reported, upon 1,25VD treatment, an apparent lack of VDR-RXR nuclear translocation in myotubes compared to myoblasts [
19]. Since proper VDR function is required for 1,25VD effects on myoblasts mitochondrial activity [
15], this could be a further mechanism underlying the differences in the effects of 1,25VD on myotubes compared to myoblasts.
On the other hand, the effects of 1,25VD upstream metabolites VD3 and 25VD on mitochondria are less straightforward. As both VD3 and 25VD own a protective activity on C2C12 myotubes in the presence of atrophic stimuli [
18,
19], it is possible to envision that more striking effects on mitochondrial metabolism could be detected in an atrophic context. The differences between VD3 and 25VD in some assays (
Figure 1b and
Figure 2b,c) may point to the fact that VD3 is not converted to 25VD in C2C12 myotubes, and that it probably acts through different mechanisms [
19]. Nevertheless, both VD3 and 25VD increased mitochondrial content (
Figure 4a) and reduced basal ROS (
Figure 6a,b), suggesting that their anti-atrophic properties can depend, in part, on antioxidant activity.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







