
Mitochondrial-Targeted Antioxidant MitoQ-Mediated Autophagy: A Novel Strategy for Precise Radiation Protection


Abstract
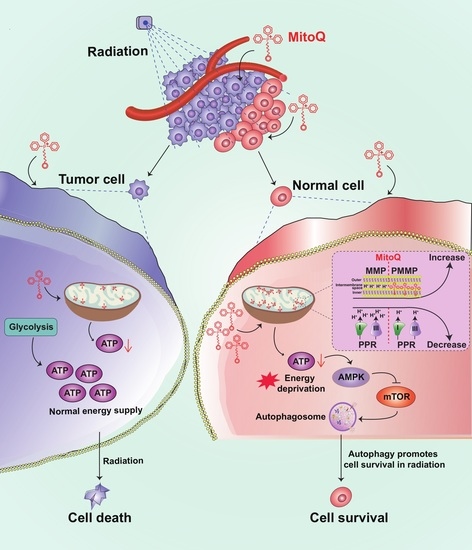
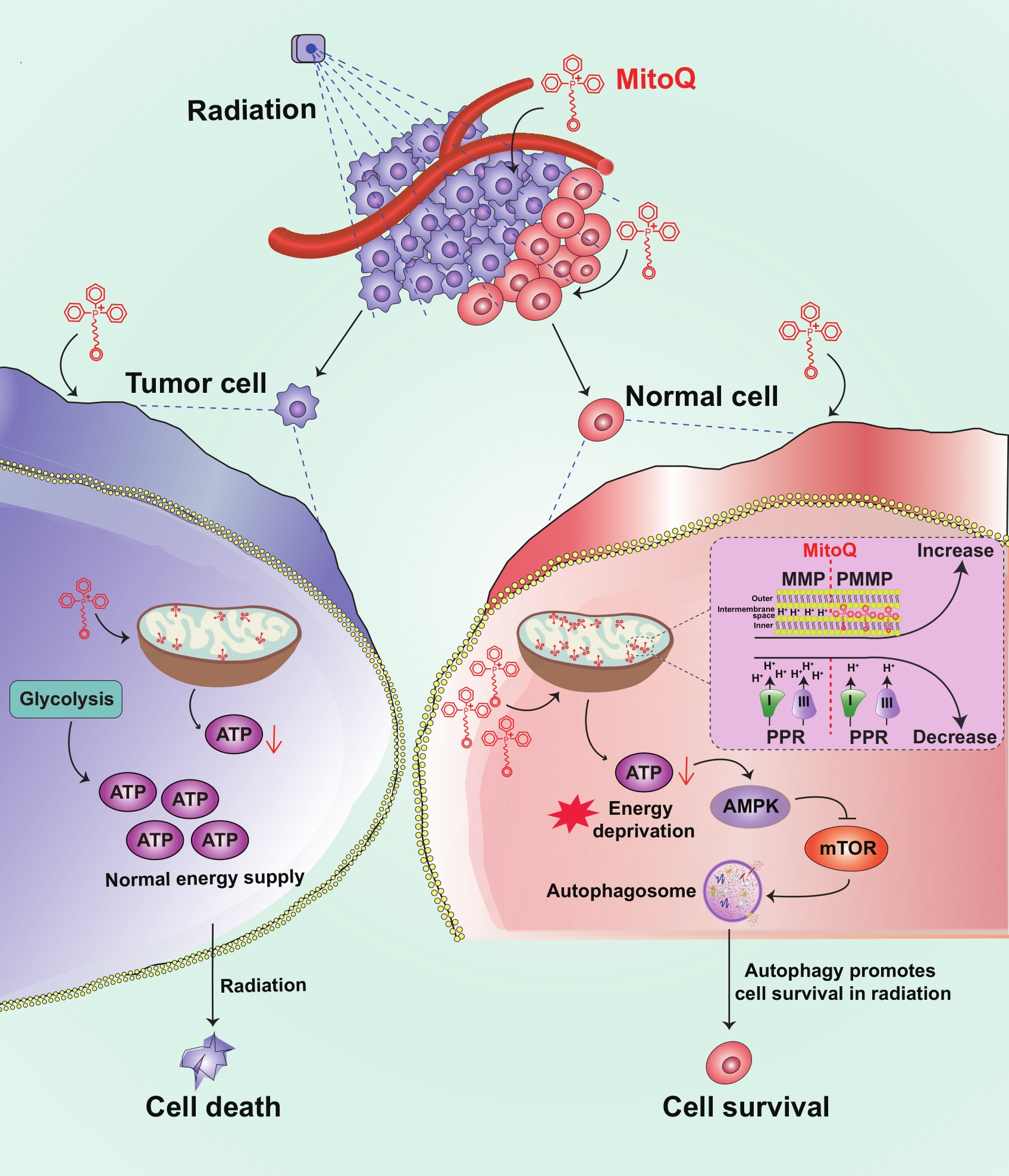
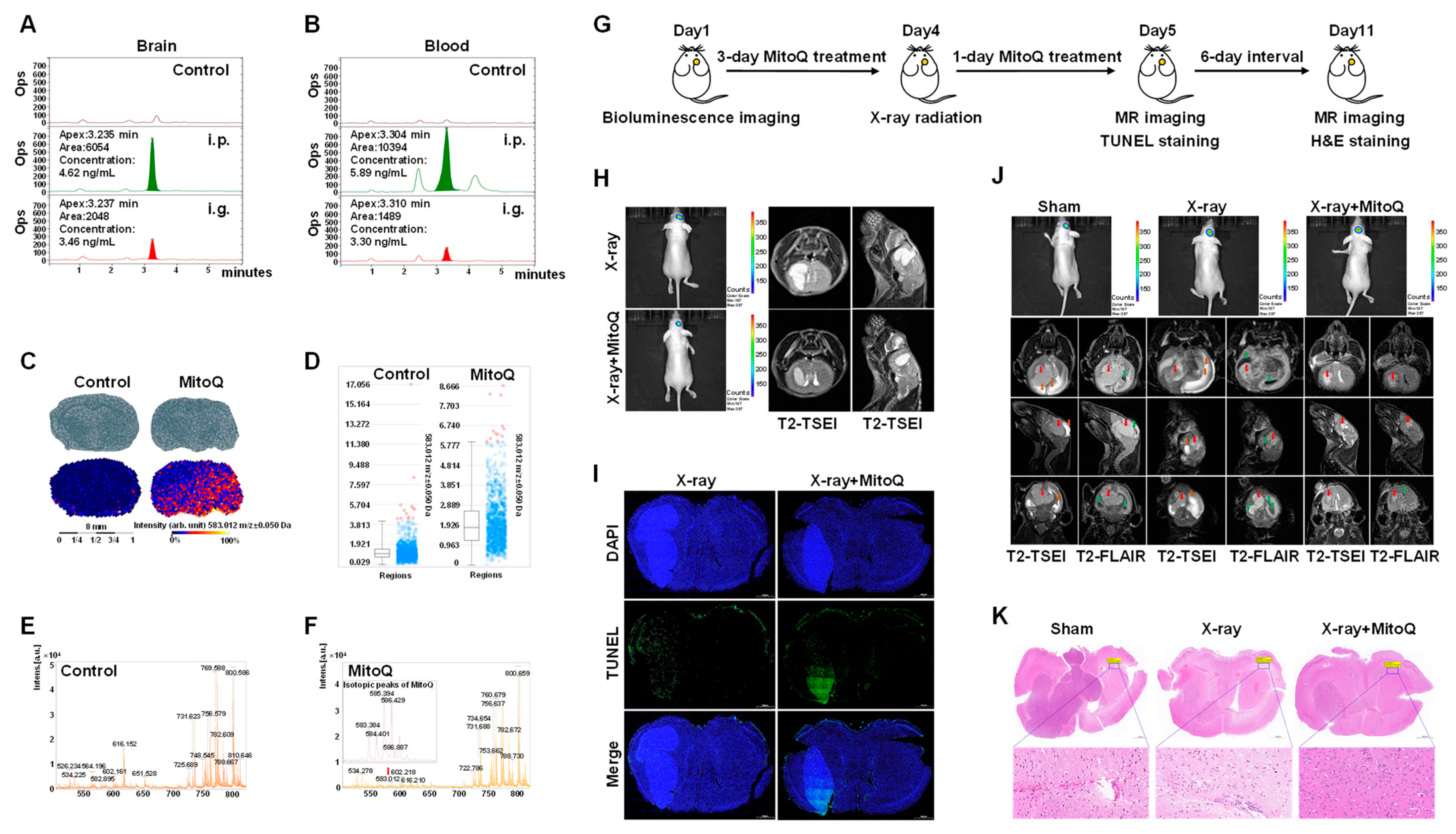
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods and Materials
2.1. Cell Culture
2.2. Animals
2.3. Polymerase Chain Reaction (PCR) Array Expression Profiling
2.4. Agilent Seahorse XF Technology
2.5. HPLC-MS Analysis
2.6. Determination of Mitochondrial Membrane Potential (MMP)
2.7. Detection of Respiratory Chain Complex Activities
2.8. Measurement of H+/2e
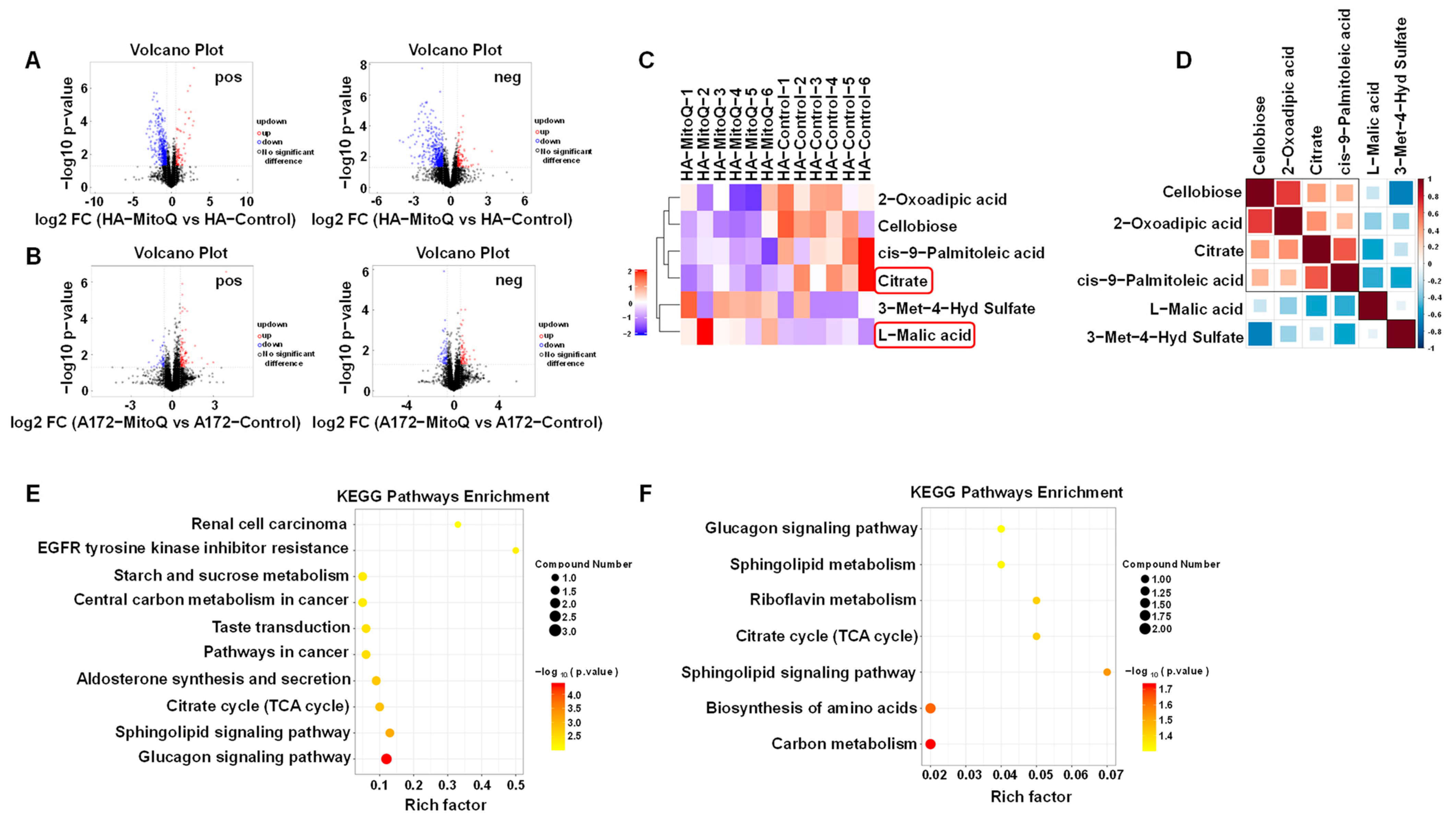
2.9. Untargeted Metabolomics Based on Ultra-High Performance Liquid Chromatography-Tandem Time-of-Flight Mass Spectrometry (UHPLC-Q-TOF MS)
2.10. Western Blot Analysis
2.11. Transmission Electron Microscopy (TEM)
2.12. Observation of Autophagosome and Autophagy Flux
2.13. In Vitro Cell Proliferation Analysis
2.14. MALDI-TOF-MS Imaging Mass Spectrometry Analysis
2.15. In Vivo Imaging of Orthotopic Glioma Using Bioluminescence Imaging (BLI) and Magnetic Resonance Imaging (MRI)
2.16. Histomorphological and Terminal dUTP Nicked End LABELING (TUNEL) Assessments of Brain
2.17. Statistical Analysis
3. Results
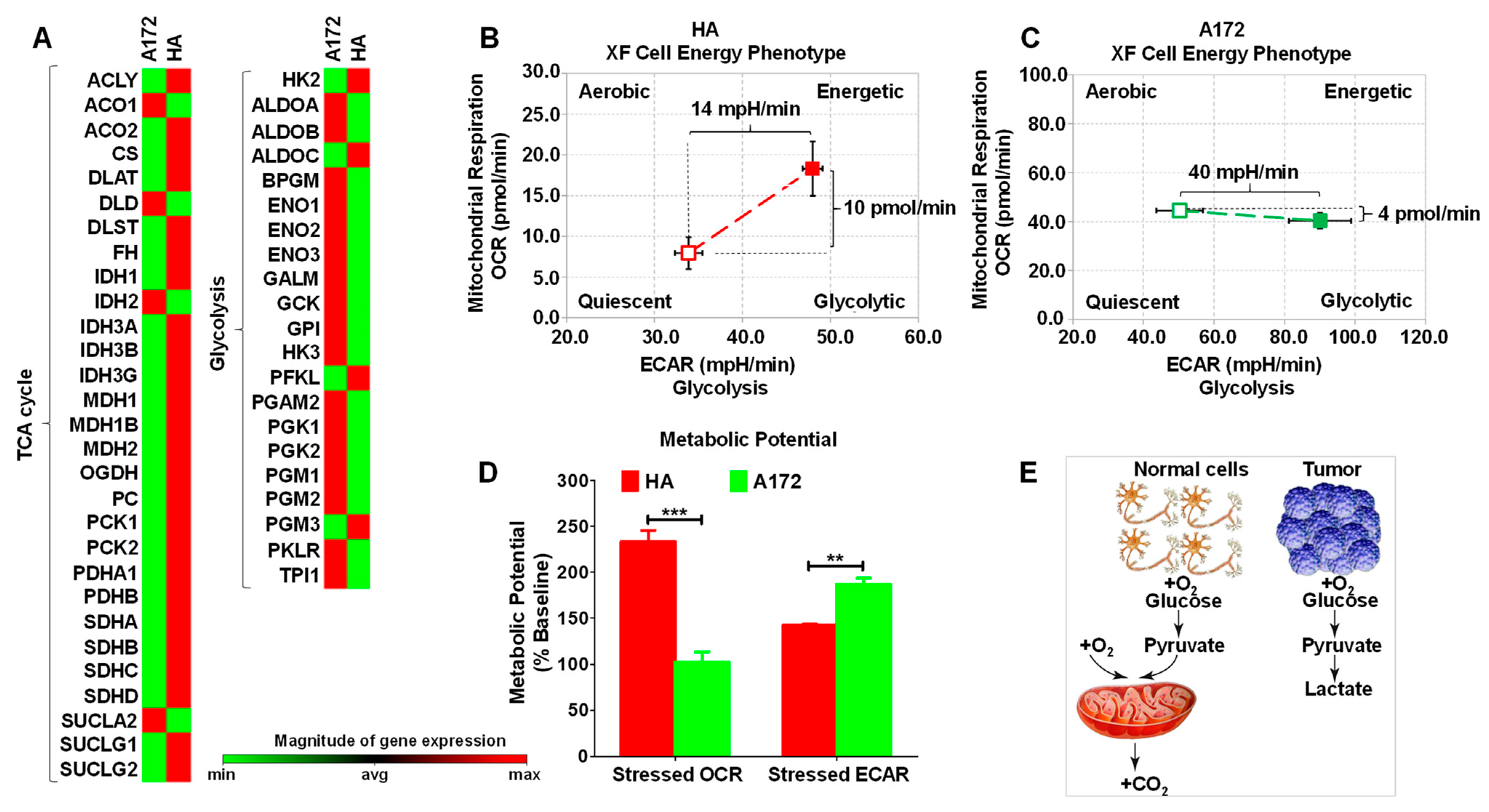
3.1. Cell Energy Phenotype of HA and A172 Cells
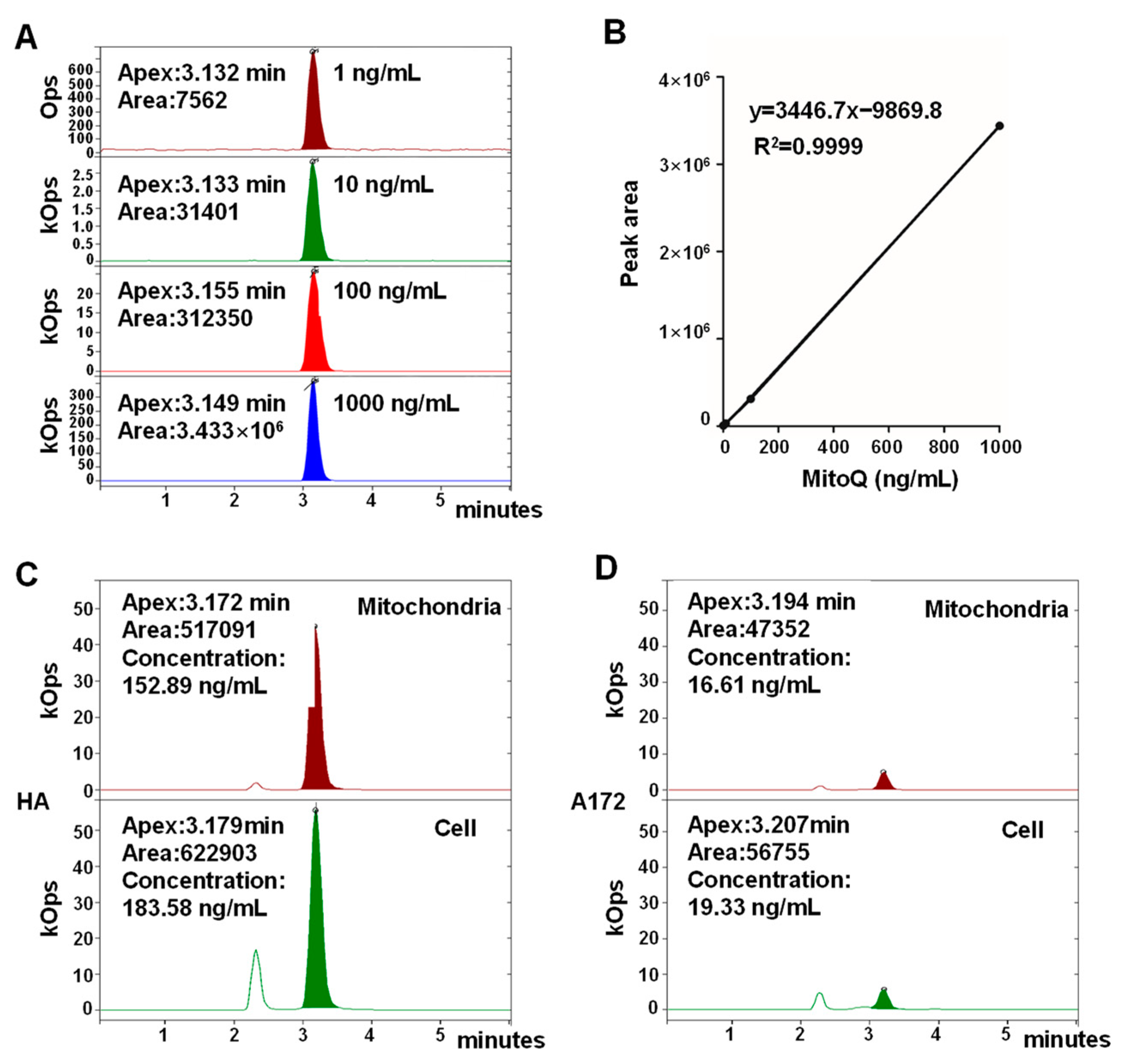
3.2. MitoQ Was Selectively Enriched in Mitochondria in HA Cells Higher than That in A172 Cells
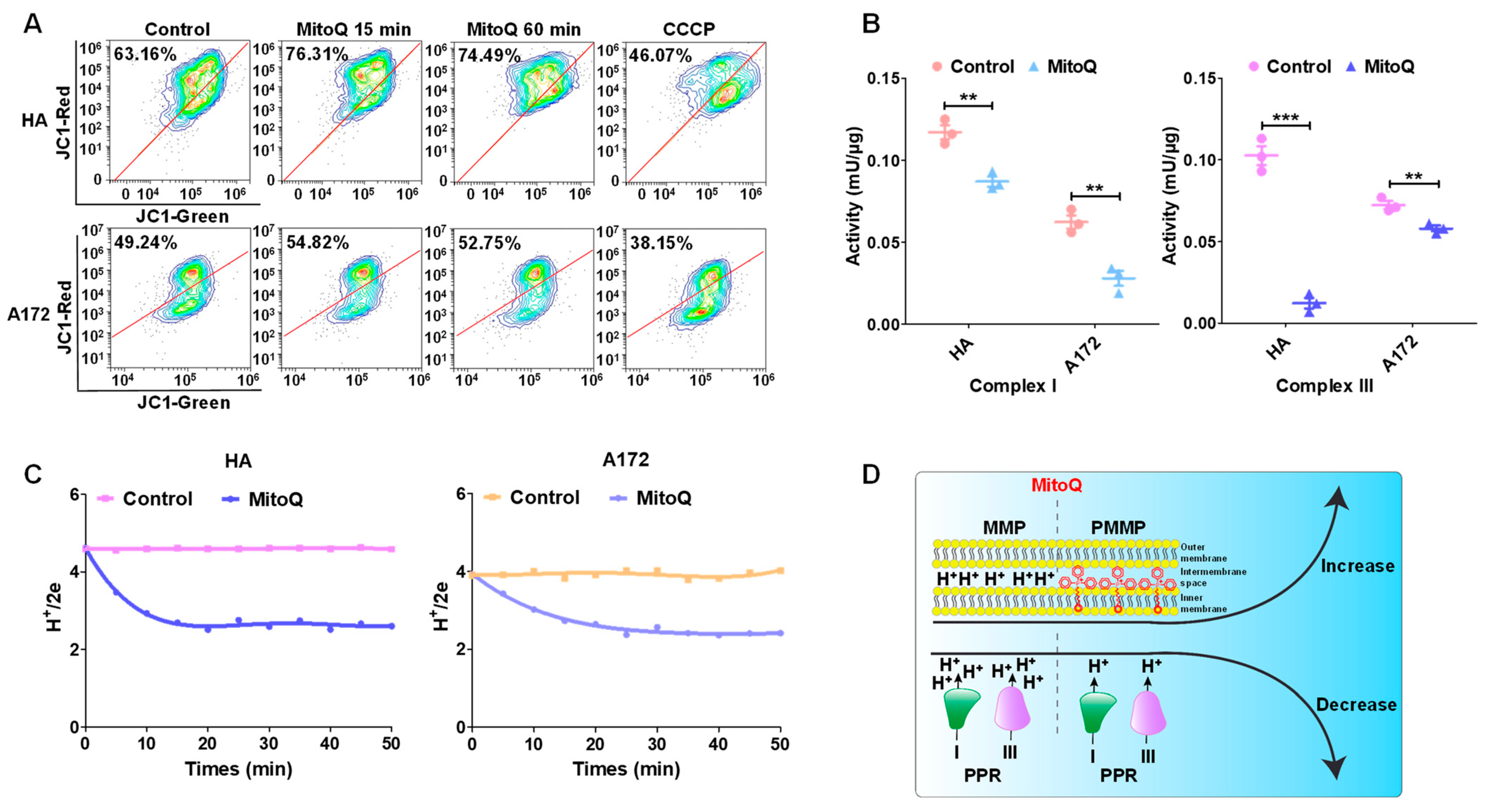
3.3. MitoQ-Induced PMMP in Normal Cells Was Higher than That in Tumor Cells
3.4. PMMP Disrupted Energy Metabolism in HA Cells
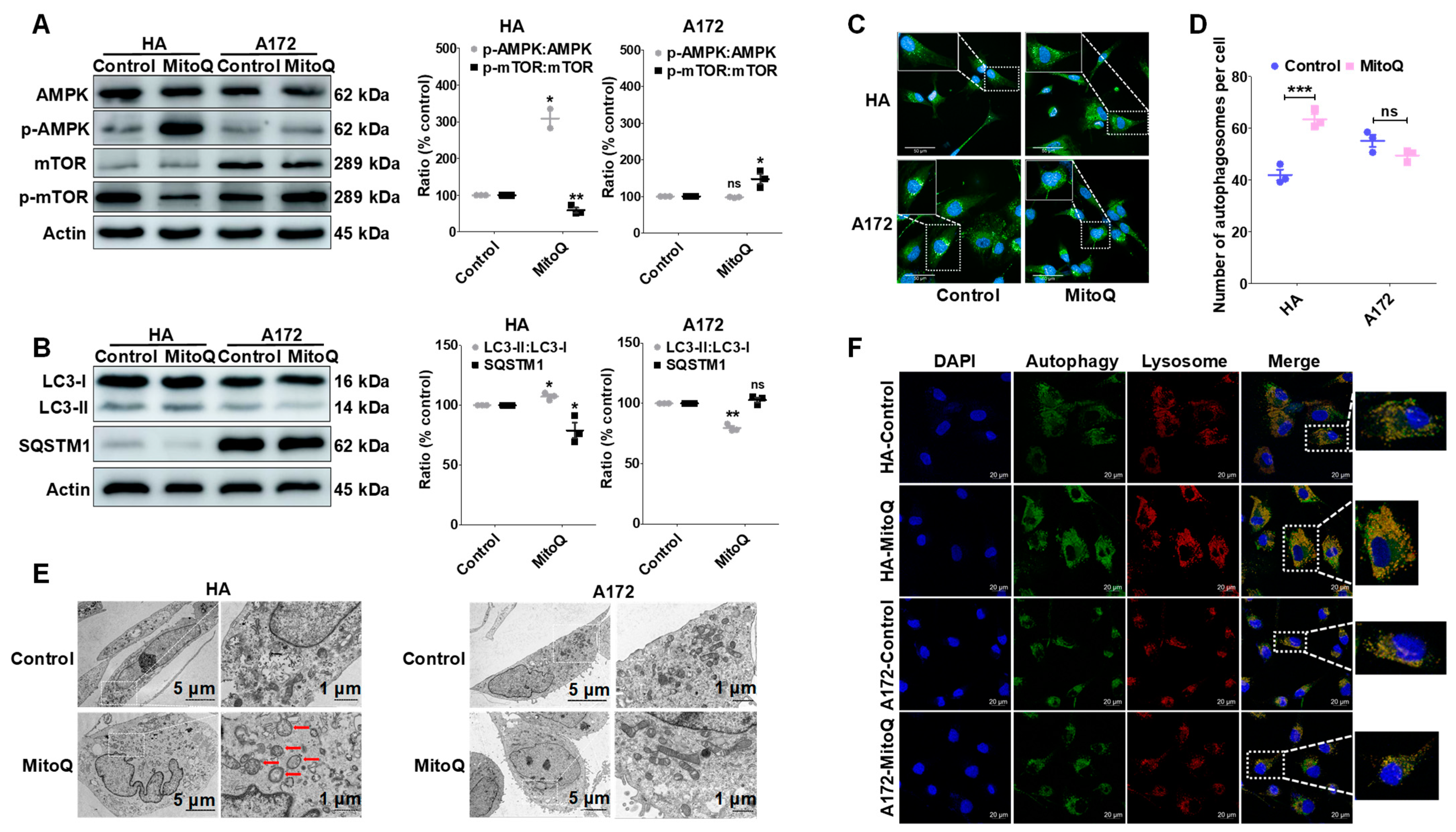
3.5. MitoQ Induced Autophagy in HA Cells through the AMPK/mTOR Pathway
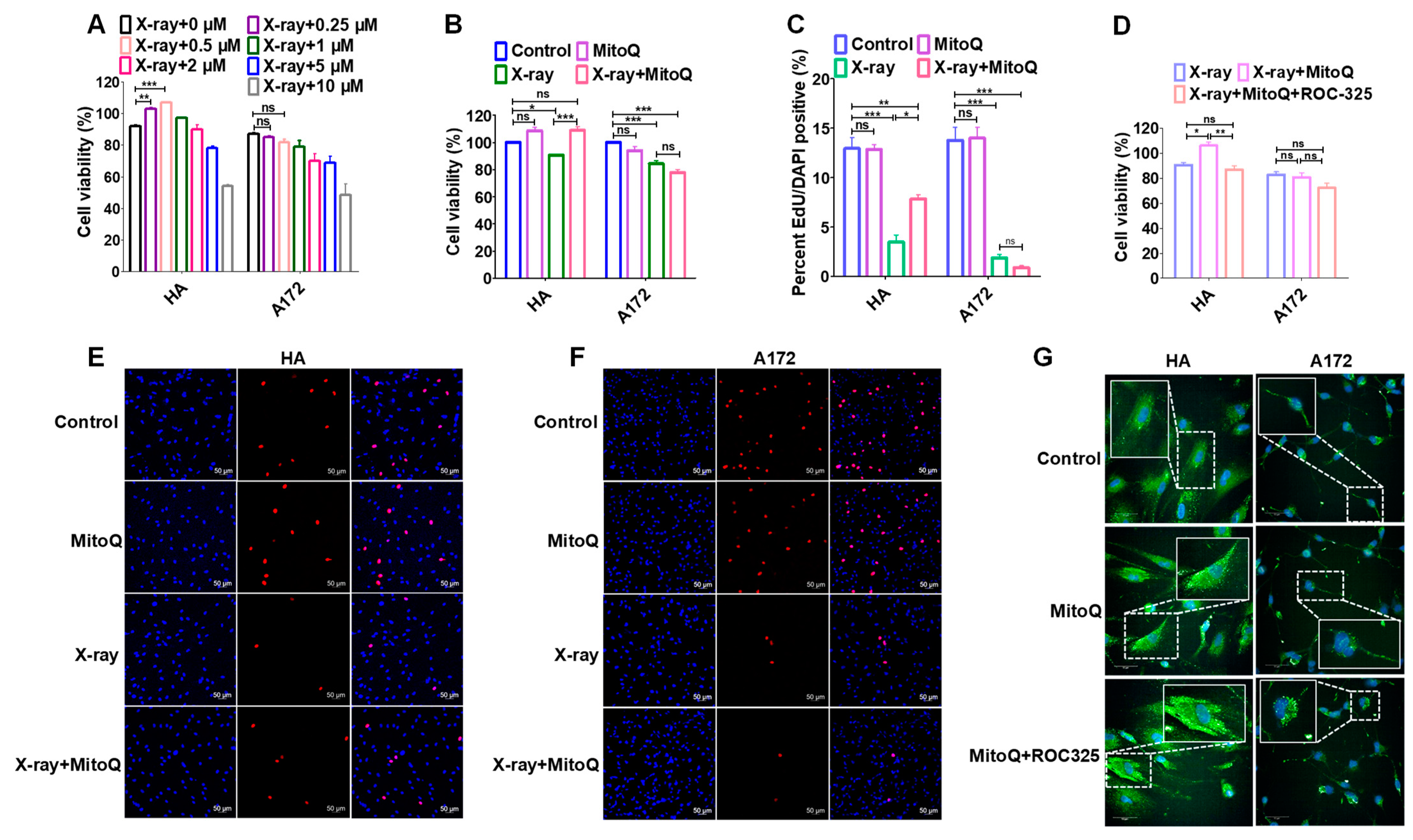
3.6. Protective Effect of MitoQ on Normal Cells against Radiation
3.7. MitoQ Protected Normal Tissue against X-rays in Mice Bearing Orthotopic Glioma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamblin, R.; Vardon, A.; Akpalu, J.; Tampourlou, M.; Spiliotis, I.; Sbardella, E.; Lynch, J.; Shankaran, V.; Mavilakandy, A.; Gagliardi, I.; et al. Risk of second brain tumour after radiotherapy for pituitary adenoma or craniopharyngioma: A retrospective, multicentre, cohort study of 3679 patients with long-term imaging surveillance. Lancet Diabetes Endocrinol. 2022, 10, 581–588. [Google Scholar] [CrossRef]
- Hong, Z.; Liu, T.; Wan, L.; Fa, P.; Kumar, P.; Cao, Y.; Prasad, B.; Qiu, Z.; Liu, J.; Wang, H.; et al. Targeting Squalene Epoxidase Interrupts Homologous Recombination via the ER Stress Response and Promotes Radiotherapy Efficacy. Cancer Res. 2022, 82, 1298–1312. [Google Scholar] [CrossRef]
- Kim, S.H.; Lim, K.H.; Yang, S.; Joo, J.Y. Long non-coding RNAs in brain tumors: Roles and potential as therapeutic targets. J. Hematol. Oncol. 2021, 14, 77–94. [Google Scholar] [CrossRef]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.M.; Hegi-Johnson, F. Radiotherapy toxicity. Nat. Rev. Dis. Prim. 2019, 5, 13–33. [Google Scholar] [CrossRef]
- ClinicalTrials.gov: A Database of Privately and Publicly Funded Clinical Studies Conducted around the World. Available online: https://clinicaltrials.gov/ct2/results?cond=Radiation+Protection+and+cancer%09&term=&cntry=&state=&city=&dist= (accessed on 13 December 2022).
- Ashcraft, K.A.; Boss, M.K.; Tovmasyan, A.; Roy Choudhury, K.; Fontanella, A.N.; Young, K.H.; Palmer, G.M.; Birer, S.R.; Landon, C.D.; Park, W.; et al. Novel Manganese-Porphyrin Superoxide Dismutase-Mimetic Widens the Therapeutic Margin in a Preclinical Head and Neck Cancer Model. Int. J. Radiat. Oncol. 2015, 93, 892–900. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Fischer, N.; Seo, E.J.; Efferth, T. Prevention from radiation damage by natural products. Phytomedicine 2018, 47, 192–200. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345. [Google Scholar] [CrossRef]
- Ismail, H.; Shakkour, Z.; Tabet, M.; Abdelhady, S.; Kobaisi, A.; Abedi, R.; Nasrallah, L.; Pintus, G.; Al-Dhaheri, Y.; Mondello, S.; et al. Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone. Antioxidants 2020, 9, 943. [Google Scholar] [CrossRef]
- Souza-Neto, F.V.; Islas, F.; Jiménez-González, S.; Luaces, M.; Ramchandani, B.; Romero-Miranda, A.; Delgado-Valero, B.; Roldan-Molina, E.; Saiz-Pardo, M.; Cerón-Nieto, M.; et al. Mitochondrial Oxidative Stress Promotes Cardiac Remodeling in Myocardial Infarction through the Activation of Endoplasmic Reticulum Stress. Antioxidants 2022, 11, 1232. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Liu, X.; Di, C.; Wang, Z.; Mi, X.; Liu, Y.; Zhao, Q.; Mao, A.; Chen, W.; Gan, L.; et al. MitoQ regulates autophagy by inducing a pseudo-mitochondrial membrane potential. Autophagy 2017, 13, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Bravo-San Pedro, J.M. Targeting Autophagy to Counteract Obesity-Associated Oxidative Stress. Antioxidants 2021, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663. [Google Scholar] [CrossRef]
- Su, K.H.; Dai, S.; Tang, Z.; Xu, M.; Dai, C. Heat Shock Factor 1 Is a Direct Antagonist of AMP-Activated Protein Kinase. Mol. Cell 2019, 76, 546–561. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, Y.; Zhou, L.; Chen, H.; Zhou, Z. From Intestinal Epithelial Homeostasis to Colorectal Cancer: Autophagy Regulation in Cellular Stress. Antioxidants 2022, 11, 1308. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Perlmutter, D.H. Autophagic disposal of the aggregation-prone protein that causes liver inflammation and carcinogenesis in alpha-1-antitrypsin deficiency. Cell Death Differ. 2009, 16, 39–45. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Rumyantseva, A.; Popovic, M.; Trifunovic, A. CLPP deficiency ameliorates neurodegeneration caused by impaired mitochondrial protein synthesis. Brain 2022, 145, 92–104. [Google Scholar] [CrossRef]
- Luo, Y.; Ma, J.; Lu, W. The Significance of Mitochondrial Dysfunction in Cancer. Int. J. Mol. Sci. 2020, 21, 5598. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Riester, M.; Xu, Q.; Moreira, A.; Zheng, J.; Michor, F.; Downey, R.J. The Warburg effect: Persistence of stem-cell metabolism in cancers as a failure of differentiation. Ann. Oncol. 2018, 29, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ibekwe, E.; Chornenkyy, Y. Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change. Antioxidants 2018, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Schulz, S.; Becker, M.; Groseclose, M.R.; Schadt, S.; Hopf, C. Advanced MALDI mass spectrometry imaging in pharmaceutical research and drug development. Curr. Opin. Biotech. 2019, 55, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cao, S.; Arhatte, M.; Li, D.; Shi, Y.; Kurz, S.; Hu, J.; Wang, L.; Shao, J.; Atzberger, A.; et al. Adipocyte Piezo1 mediates obesogenic adipogenesis through the FGF1/FGFR1 signaling pathway in mice. Nat. Commun. 2020, 11, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, L.; Wang, J.; Wang, J.; Wang, L.; Wang, R.; Wang, R.; Li, R.; Liu, H.; Wei, S.; et al. Therapeutic effects of higenamine combined with [6]-gingerol on chronic heart failure induced by doxorubicin via ameliorating mitochondrial function. J. Cell. Mol. Med. 2020, 24, 4036–4050. [Google Scholar] [CrossRef]
- Oh, S.; Yeom, J.; Cho, H.J.; Kim, J.H.; Yoon, S.J.; Kim, H.; Sa, J.K.; Ju, S.; Lee, H.; Oh, M.J.; et al. Integrated pharmaco-proteogenomics defines two subgroups in isocitrate dehydrogenase wild-type glioblastoma with prognostic and therapeutic opportunities. Nat. Commun. 2020, 11, 3288–3304. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the basics of cancer metabolism: Developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol. 2017, 12, 833–842. [Google Scholar] [CrossRef]
- Battogtokh, G.; Choi, Y.S.; Kang, D.S.; Park, S.J.; Shim, M.S.; Huh, K.M.; Cho, Y.-Y.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: Current strategies and future perspectives. Acta Pharm. Sin. B 2018, 8, 862–880. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674–101683. [Google Scholar] [CrossRef] [PubMed]
- Bauermeister, A.; Mannochio-Russo, H.; Costa-Lotufo, L.V.; Jarmusch, A.K.; Dorrestein, P.C. Mass spectrometry-based metabolomics in microbiome investigations. Nat. Rev. Microbiol. 2022, 20, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Guo, J.Y.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, H.; Zhang, L.; Lin, X.; Li, X.; Zhuang, H.; Fan, H.; Meng, T.; He, Z.; Huang, H.; et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 2021, 17, 1142–1156. [Google Scholar] [CrossRef]
- Luo, R.; Fan, Y.; Yang, J.; Ye, M.; Zhang, D.F.; Guo, K.; Li, X.; Bi, R.; Xu, N.; Yang, L.-X.; et al. A novel missense variant in ACAA1 contributes to early-onset Alzheimer’s disease, impairs lysosomal function, and facilitates amyloid-beta pathology and cognitive decline. Signal Transduct. Target. Ther. 2021, 6, 325–341. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Wang, F.; Xiao, Y.; Yan, J.-F.; Huang, G.-M.; Zhang, J.-H.; Di, C.-X.; Si, J.; Zhang, H.; Mao, A.-H. Carbon ion irradiation-induced DNA damage evokes cell cycle arrest and apoptosis via the pRb/E2F1/c-Myc signaling pathway in p53-deficient prostate cancer PC-3 cells. Nucl. Sci. Tech. 2021, 32, 30–42. [Google Scholar] [CrossRef]
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Han, Y.; Visconte, V.; Phillips, J.; Nawrocki, S.T. Disruption of Autophagic Degradation with ROC-325 Antagonizes Renal Cell Carcinoma Pathogenesis. Clin. Cancer Res. 2017, 23, 2869–2879. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.M.; Wang, X.Y.; Zhou, X.R.; Zhang, C.; Liu, K.J.; Zhang, F.Y.; Xiang, B. Salidroside Ameliorates Radiation Damage by Reducing Mitochondrial Oxidative Stress in the Submandibular Gland. Antioxidants 2022, 11, 1414. [Google Scholar] [CrossRef] [PubMed]
- Chauveinc, L.; Giraud PFau-Dahnier, S.; Dahnier SFau-Mounier, N.; Mounier NFau-Cosset, J.M.; Cosset, J.M. Radiotherapy-induced solid tumors: Review of the literature and risk assessment. Cancer Radiother. 1998, 2, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Rubino, C.; de Vathaire, F.; Shamsaldin, A.; Labbe, M.; Le, M.G. Radiation dose, chemotherapy, hormonal treatment and risk of second cancer after breast cancer treatment. Brit. J. Cancer 2003, 89, 840–846. [Google Scholar] [CrossRef]
- Dorr, W.; Herrmann, T. Second tumors after oncologic treatment. Strahlenther. Onkol. 2008, 184, 67–72. [Google Scholar] [CrossRef]
- Huh, J.W.; Tanksley, J.; Chino, J.; Willett, C.G.; Dewhirst, M.W. Long-term Consequences of Pelvic Irradiation: Toxicities, Challenges, and Therapeutic Opportunities with Pharmacologic Mitigators. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3079–3090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, K.; Zhang, Q.; Zhu, Z.; Huang, G.; Tian, H. Polycysteine as a new type of radio-protector ameliorated tissue injury through inhibiting ferroptosis in mice. Cell Death Dis. 2021, 12, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhang, H.; Liu, H.; Han, Y.; Qiu, W.; Li, Z. Inhibiting autophagy flux and DNA repair of tumor cells to boost radiotherapy of orthotopic glioblastoma. Biomaterials 2022, 280, 121287–121299. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zeng, L.; Su, P.; Ma, L.; Zhang, M.; Zhang, Y.Z. Autophagy: A multifaceted player in the fate of sperm. Hum. Reprod. Update 2022, 28, 200–231. [Google Scholar] [CrossRef]
- Kim, D.; Kim, J.; Yu, Y.S.; Kim, Y.R.; Baek, S.H.; Won, K.J. Systemic approaches using single cell transcriptome reveal that C/EBPgamma regulates autophagy under amino acid starved condition. Nucleic Acids Res. 2022, 50, 7298–7309. [Google Scholar] [CrossRef]
- Ruart, M.; Chavarria, L.; Camprecios, G.; Suarez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef]
- Park, S.J.; Frake, R.A.; Karabiyik, C.; Son, S.M.; Siddiqi, F.H.; Bento, C.F.; Sterk, P.; Vicinanza, M.; Pavel, M.; Rubinsztein, D.C. Vinexin contributes to autophagic decline in brain ageing across species. Cell Death Differ. 2022, 29, 1055–1070. [Google Scholar] [CrossRef]
- Jing, Z.; Liu, Q.; He, X.; Jia, Z.; Xu, Z.; Yang, B.; Liu, P. NCAPD3 enhances Warburg effect through c-myc and E2F1 and promotes the occurrence and progression of colorectal cancer. J. Exp. Clin. Cancer Res. 2022, 41, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sun, Z.; Liang, M.; Song, W.; Zhang, R.; Shi, Y.; Cui, Y.; Gao, Q. An Unrevealed Molecular Function of Corannulene Buckybowl Glycoconjugates in Selective Tumor Annihilation by Targeting the Cancer-Specific Warburg Effect. Adv. Sci. 2022, 9, 2105315–2105330. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ge, Y.; Dong, J.; Wang, H.; Zhao, T.; Wang, X.; Liu, J.; Gao, S.; Shi, L.; Yang, S.; et al. BZW1 Facilitates Glycolysis and Promotes Tumor Growth in Pancreatic Ductal Adenocarcinoma Through Potentiating eIF2alpha Phosphorylation. Gastroenterology 2022, 162, 1256–1271. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, X.; Liu, X.; Wu, Q.; Ye, F.; Shi, Z.; Xu, D.; Zhang, J.; Dou, Z.; Huang, G.; Zhang, H.; et al. Mitochondrial-Targeted Antioxidant MitoQ-Mediated Autophagy: A Novel Strategy for Precise Radiation Protection. Antioxidants 2023, 12, 453. https://doi.org/10.3390/antiox12020453
Bao X, Liu X, Wu Q, Ye F, Shi Z, Xu D, Zhang J, Dou Z, Huang G, Zhang H, et al. Mitochondrial-Targeted Antioxidant MitoQ-Mediated Autophagy: A Novel Strategy for Precise Radiation Protection. Antioxidants. 2023; 12(2):453. https://doi.org/10.3390/antiox12020453
Chicago/Turabian StyleBao, Xingting, Xiongxiong Liu, Qingfeng Wu, Fei Ye, Zheng Shi, Dan Xu, Jinhua Zhang, Zhihui Dou, Guomin Huang, Hong Zhang, and et al. 2023. "Mitochondrial-Targeted Antioxidant MitoQ-Mediated Autophagy: A Novel Strategy for Precise Radiation Protection" Antioxidants 12, no. 2: 453. https://doi.org/10.3390/antiox12020453
APA StyleBao, X., Liu, X., Wu, Q., Ye, F., Shi, Z., Xu, D., Zhang, J., Dou, Z., Huang, G., Zhang, H., & Sun, C. (2023). Mitochondrial-Targeted Antioxidant MitoQ-Mediated Autophagy: A Novel Strategy for Precise Radiation Protection. Antioxidants, 12(2), 453. https://doi.org/10.3390/antiox12020453