

Phosphatidylcholine Liposomes Reprogram Macrophages toward an Inflammatory Phenotype


Abstract
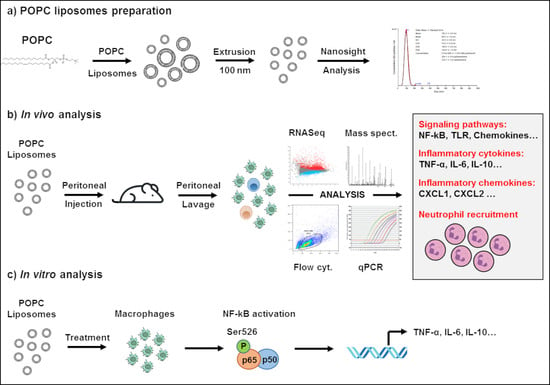
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Isolation and Culture
2.3. Liposome Preparation and Treatment
2.4. RNA Extraction, cDNA Production and Quantitative Real-Time PCR (qPCR)
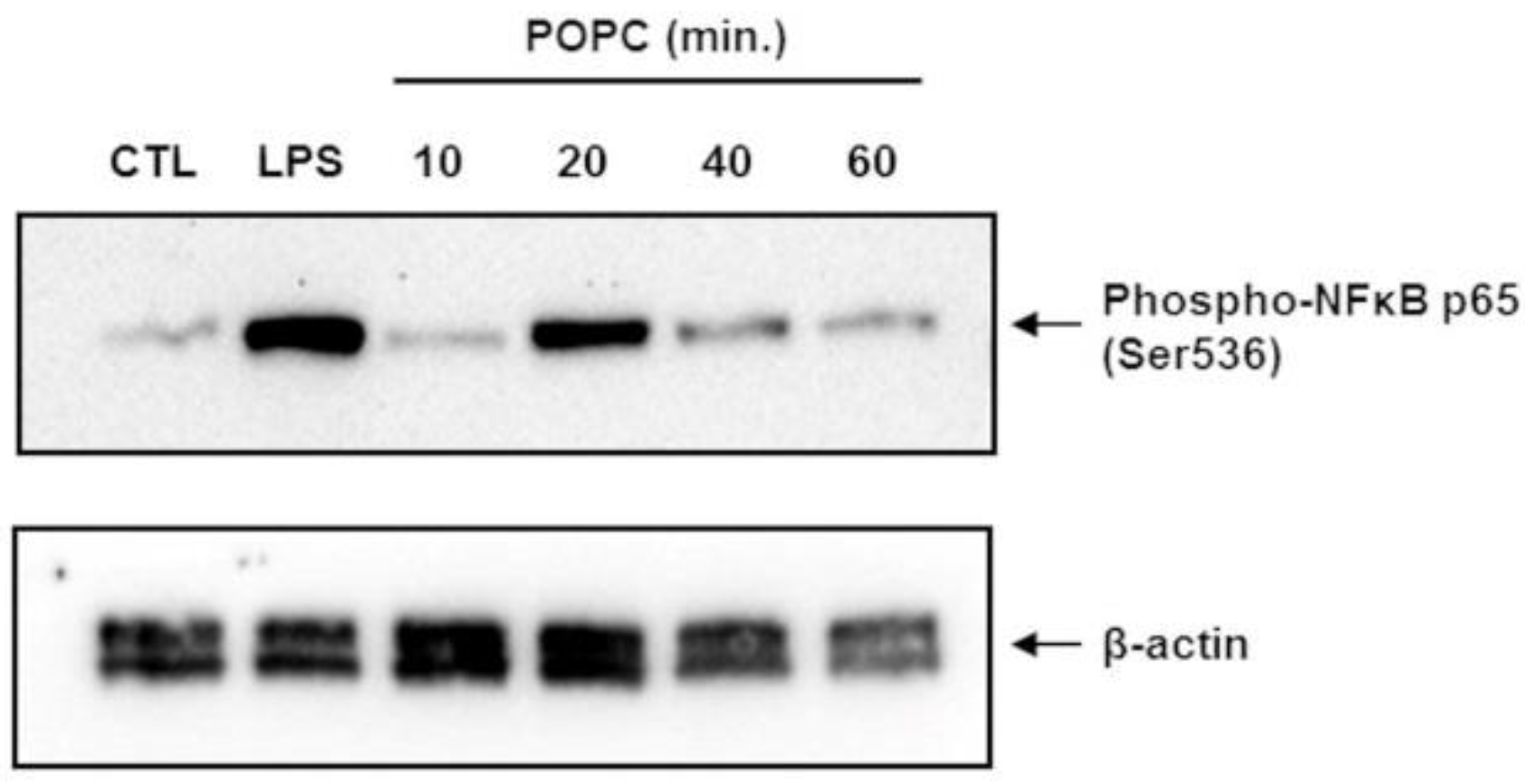
2.5. Immunoblot Analysis
2.6. Global Gene Analysis by RNA Sequencing
2.7. Flow Cytometry Analysis of Peritoneal Cells
2.8. POPC Liposome Uptake
2.9. Mass Spectrometry Analysis
2.10. Statistical Analysis
3. Results
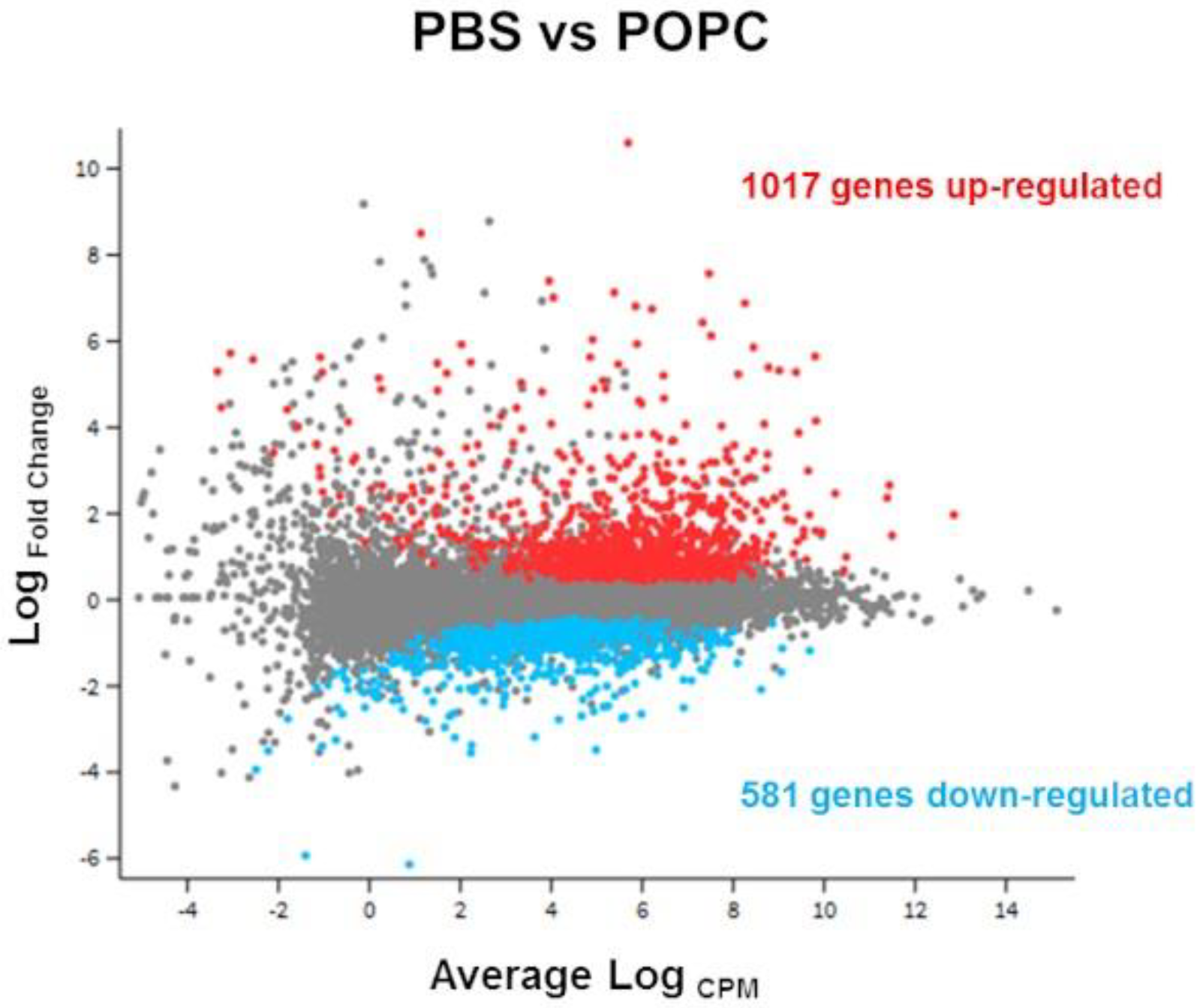
3.1. POPC Liposomes Trigger a Robust Inflammatory Response in Macrophages
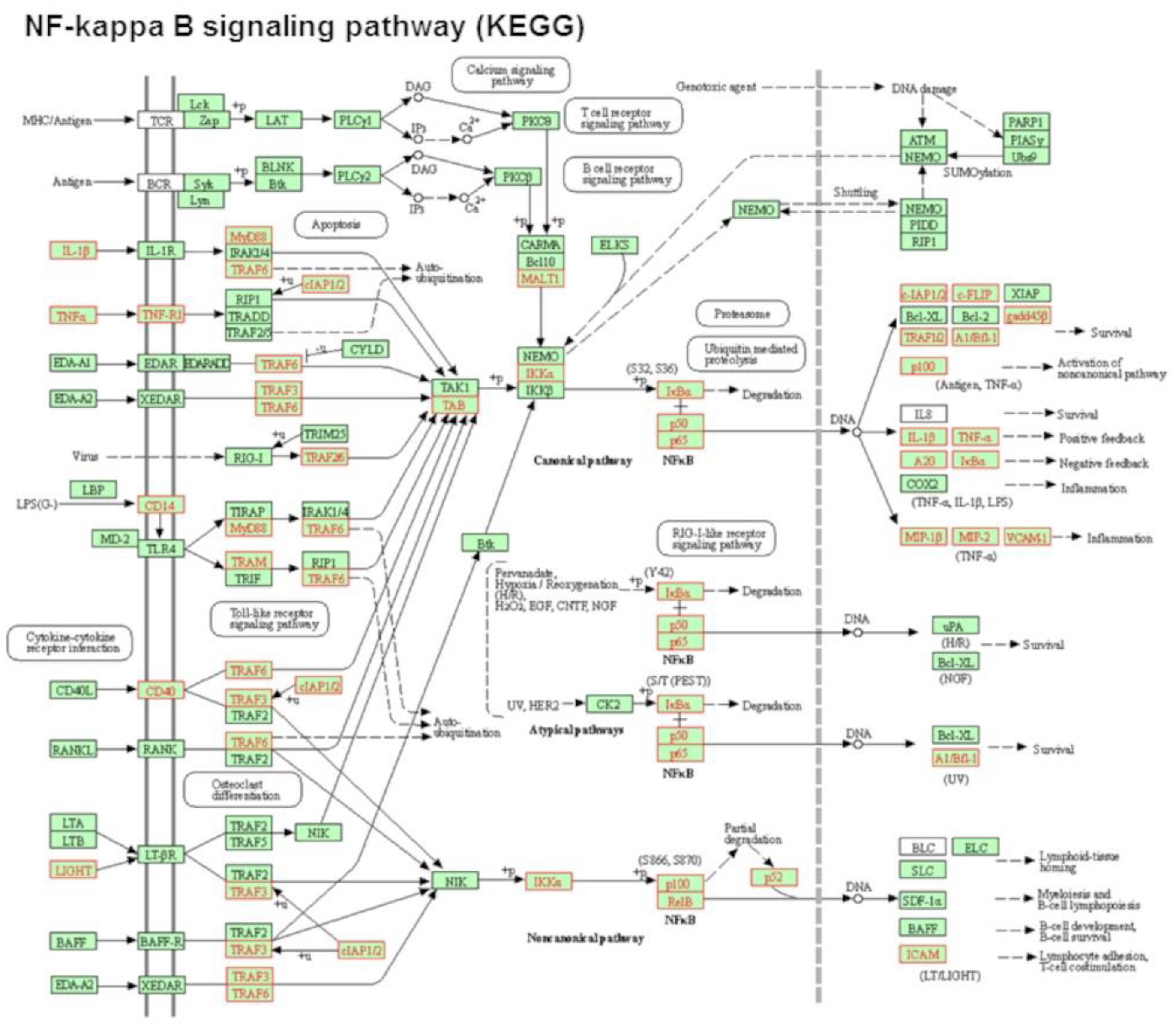
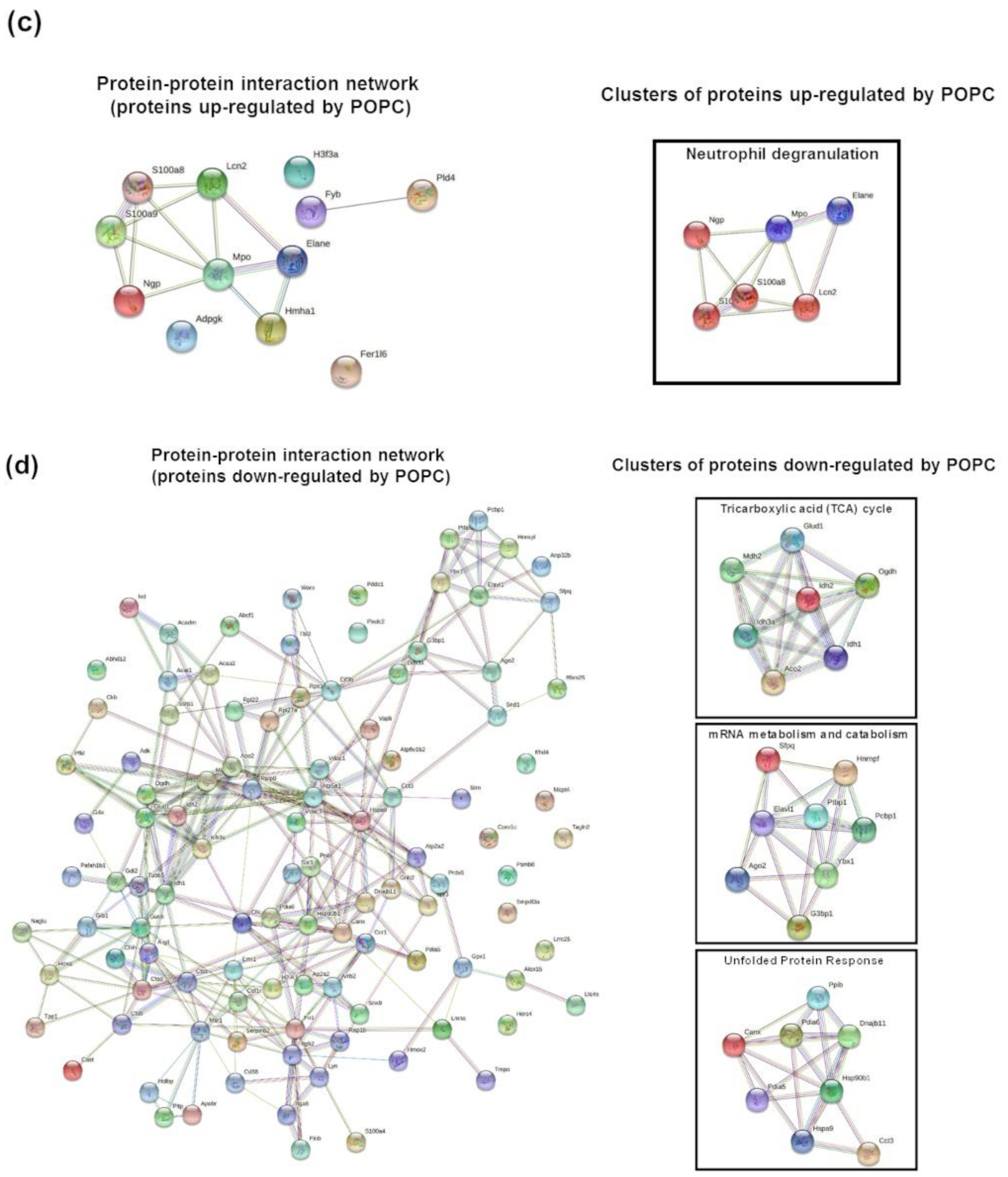
3.2. POPC Liposomes Induce Changes in the Proteome of Peritoneal Cells
3.3. POPC Liposomes Elicit the Rapid Recruitment of Neutrophils into the Peritoneal Cavity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids. 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Peng, Y.; Xu, H.; Cui, Z.; Williams, R.O., 3rd. The COVID-19 Vaccine Race: Challenges and Opportunities in Vaccine Formulation. AAPS PharmSciTech. 2020, 21, 225. [Google Scholar] [CrossRef] [PubMed]
- Heaton, P.M. The Covid-19 Vaccine-Development Multiverse. N. Engl. J. Med. 2020, 383, 1986–1988. [Google Scholar] [CrossRef]
- Calina, D.; Hernandez, A.F.; Hartung, T.; Egorov, E.M.; Izotov, B.N.; Nikolouzakis, T.K.; Tsatsakis, A.; Vlachoyiannopoulos, P.G.; Docea, A.O. Challenges and Scientific Prospects of the Newest Generation of mRNA-Based Vaccines against SARS-CoV-2. Life 2021, 11, 907. [Google Scholar] [CrossRef]
- Tanford, C. The hydrophobic effect and the organization of living matter. Science 1978, 200, 1012–1018. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- van Rooijen, N.; van Nieuwmegen, R. Liposomes in immunology: Multilamellar phosphatidylcholine liposomes as a simple, biodegradable and harmless adjuvant without any immunogenic activity of its own. Immunol. Commun. 1980, 9, 243–256. [Google Scholar] [CrossRef]
- Furse, S.; de Kroon, A.I. Phosphatidylcholine’s functions beyond that of a membrane brick. Mol. Membr. Biol. 2015, 32, 117–119. [Google Scholar] [CrossRef]
- Cauvi, D.M.; Hawisher, D.; Dores-Silva, P.R.; Lizardo, R.E.; De Maio, A. Macrophage reprogramming by negatively charged membrane phospholipids controls infection. FASEB J. 2019, 33, 2995–3009. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Zhang, K.; Hendrie, C.; Liang, C.; Li, M.; Doherty-Kirby, A.; Lajoie, G. PEAKS: Powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2337–2342. [Google Scholar] [CrossRef] [PubMed]
- Casares, D.; Escriba, P.V.; Rossello, C.A. Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues. Int. J. Mol. Sci. 2019, 20, 2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Lo Cicero, A.; Stahl, P.D.; Raposo, G. Extracellular vesicles shuffling intercellular messages: For good or for bad. Curr. Opin. Cell Biol. 2015, 35, 69–77. [Google Scholar] [CrossRef]
- De Maio, A. Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: A form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell Stress Chaperones. 2011, 16, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, J.; Larrick, J.W.; Ryan, J.L.; Szoka, F.C. Incorporation of LPS in liposomes diminishes its ability to induce tumoricidal activity and tumor necrosis factor secretion in murine macrophages. J. Leukoc. Biol. 1998, 43, 436–444. [Google Scholar] [CrossRef]
- Exton, J.H. Phosphatidylcholine breakdown and signal transduction. Biochim. Biophys. Acta. 1994, 1212, 26–42. [Google Scholar] [CrossRef]
- Huang, Y.H.; Schafer-Elinder, L.; Wu, R.; Claesson, H.E.; Frostegard, J. Lysophosphatidylcholine (LPC) induces proinflammatory cytokines by a platelet-activating factor (PAF) receptor-dependent mechanism. Clin. Exp. Immunol. 1999, 116, 326–331. [Google Scholar] [CrossRef]
- Carneiro, A.B.; Iaciura, B.M.; Nohara, L.L.; Lopes, C.D.; Veas, E.M.C.; Mariano, V.S.; Bozza, P.T.; Lopes, U.G.; Atella, G.C.; Almeida, I.C.; et al. Lysophosphatidylcholine triggers TLR2- and TLR4-mediated signaling pathways but counteracts LPS-induced NO synthesis in peritoneal macrophages by inhibiting NF-kappaB translocation and MAPK/ERK phosphorylation. PLoS One 2013, 8, e76233. [Google Scholar] [CrossRef]
- Angers, M.; Uldry, M.; Kong, D.; Gimble, J.M.; Jetten, A.M. Mfsd2a encodes a novel major facilitator superfamily domain-containing protein highly induced in brown adipose tissue during fasting and adaptive thermogenesis. Biochem J. 2008, 416, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, W.R.; Goh, E.L.; Silver, D.L. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef]
- Piccirillo, A.R.; Hyzny, E.J.; Beppu, L.Y.; Menk, A.V.; Wallace, C.T.; Hawse, W.F.; Buechel, H.M.; Wong, B.H.; Foo, J.C.; Cazenave-Gassiot, A.; et al. The Lysophosphatidylcholine Transporter MFSD2A Is Essential for CD8(+) Memory T Cell Maintenance and Secondary Response to Infection. J. Immunol. 2019, 203, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Eser Ocak, P.; Ocak, U.; Sherchan, P.; Zhang, J.H.; Tang, J. Insights into major facilitator superfamily domain-containing protein-2a (Mfsd2a) in physiology and pathophysiology. What do we know so far? J. Neurosci. Res. 2020, 98, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungaro, F.; Tacconi, C.; Massimino, L.; Corsetto, P.A.; Correale, C.; Fonteyne, P.; Piontini, A.; Garzarelli, V.; Calcaterra, F.; Della Bella, S.; et al. MFSD2A Promotes Endothelial Generation of Inflammation-Resolving Lipid Mediators and Reduces Colitis in Mice. Gastroenterology 2017, 153, 1363–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [Green Version]
- Jaishy, B.; Abel, E.D. Lipids, lysosomes, and autophagy. J. Lipid Res. 2016, 57, 1619–1635. [Google Scholar] [CrossRef] [Green Version]
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, S.; Carlson, T.; Dellacasagrande, J.; Garcia, A.; Gibbons, S.; Hertzog, P.; Lyons, A.; Lin, L.-L.; Lynch, M.; Monie, T.; et al. TRIL, a functional component of the TLR4 signaling complex, highly expressed in brain. J. Immunol. 2009, 183, 3989–3995. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, S.; Wochal, P.; Dunne, A.; O’Neill, L.A.J. Toll-like receptor 3 (TLR3) signaling requires TLR4 Interactor with leucine-rich REPeats (TRIL). J. Biol. Chem. 2011, 286, 38795–38804. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway Name | pSize | NDE | pNDE | pGFdr | Status |
|---|---|---|---|---|---|
| NF-kappa B signaling pathway | 83 | 29 | 1.49 × 10−7 | 2.81 × 10−9 | Activated |
| Osteoclast differentiation | 109 | 29 | 6.44 × 10−5 | 4.78 × 10−7 | Activated |
| Herpes simplex infection | 160 | 44 | 3.44 × 10−7 | 2.17 × 10−6 | Activated |
| MAPK signaling pathway | 199 | 40 | 1.92 × 10−3 | 5.99 × 10−6 | Activated |
| Cytokine-cytokine receptor interaction | 164 | 34 | 2.39 × 10−3 | 5.99 × 10−6 | Activated |
| Apoptosis | 76 | 24 | 1.29 × 10−5 | 2.45 × 10−5 | Activated |
| Legionellosis | 53 | 21 | 7.15 × 10−7 | 2.85 × 10−5 | Activated |
| Influenza A | 137 | 39 | 6.05 × 10−7 | 2.85 × 10−5 | Activated |
| Tuberculosis | 146 | 27 | 2.72 × 10−2 | 3.31 × 10−5 | Activated |
| Toxoplasmosis | 105 | 32 | 1.19 × 10−6 | 3.84 × 10−5 | Activated |
| Measles | 115 | 34 | 1.22 × 10−6 | 4.34 × 10−5 | Activated |
| NOD-like receptor signaling pathway | 54 | 21 | 1.03 × 10−6 | 7.28 × 10−5 | Inhibited |
| Toll-like receptor signaling pathway | 85 | 26 | 1.09 × 10−5 | 1.64 × 10−4 | Activated |
| Jak-STAT signaling pathway | 111 | 30 | 3.51 × 10−5 | 3.42 × 10−4 | Inhibited |
| Hepatitis C | 96 | 27 | 3.99 × 10−5 | 1.05 × 10−3 | Activated |
| Malaria | 39 | 14 | 1.79 × 10−4 | 2.75 × 10−3 | Activated |
| Adipocytokine signaling pathway | 52 | 16 | 4.85 × 10−4 | 3.01 × 10−3 | Activated |
| Chagas disease (American trypanosomiasis) | 93 | 23 | 1.07 × 10−3 | 3.01 × 10−3 | Activated |
| Transcriptional misregulation in cancer | 134 | 33 | 1.09 × 10−4 | 3.01 × 10−3 | Activated |
| Chemokine signaling pathway | 145 | 26 | 4.18 × 10−2 | 3.01 × 10−3 | Activated |
| Pertussis | 62 | 17 | 1.40 × 10−3 | 4.50 × 10−3 | Activated |
| Epstein-Barr virus infection | 184 | 41 | 1.91 × 10−4 | 4.84 × 10−3 | Activated |
| RIG-I-like receptor signaling pathway | 51 | 15 | 1.21 × 10−3 | 1.36 × 10−2 | Activated |
| Amyotrophic lateral sclerosis (ALS) | 40 | 8 | 1.26 × 10−1 | 1.82 × 10−2 | Activated |
| Gene Names | Gene ID | Fold Increase | Adj.p-Values |
|---|---|---|---|
| Interleukin-1 alpha | Il1a | 37 | 0.0127 |
| Interleukin-1 beta | Il1b | 107 | 0.0214 |
| Interleukin-6 | Il6 | 44 | 0.0038 |
| Interleukin-10 | Il10 | 45 | 0.0155 |
| Tumor necrosis factor-alpha | Tnfa | 86 | 0.0024 |
| Tumor necrosis factor alpha-induced protein 2 | Tnfaip2 | 50 | 0.0003 |
| C-C motif chemokine 2 | Ccl2 | 66 | 0.0257 |
| C-C motif chemokine 3 | Ccl3 | 140 | 0.0184 |
| C-C motif chemokine 4 | Ccl4 | 129 | 0.0450 |
| C-X-C motif chemokine ligand 1 | Cxcl1 | 118 | 0.0004 |
| C-X-C motif chemokine ligand 2 | Cxcl2 | 58 | 0.0004 |
| Colony Stimulating Factor 1 | Csf1 | 14 | 0.0027 |
| Colony Stimulating Factor 2 | Csf2 | 39 | 0.0372 |
| Colony Stimulating Factor 3 | Csf3 | 362 | 0.0313 |
| C-C Motif Chemokine Receptor Like 2 | Ccrl2 | 16 | 0.0015 |
| Intercellular adhesion molecule 1 | Icam1 | 39 | 0.0002 |
| Vascular cell adhesion molecule 1 | Vcam1 | 29 | 0.0201 |
| Toll like receptor 2 | Tlr2 | 17 | 0.0002 |
| Myeloid differentiation primary response 88 | Myd88 | 4 | 0.0021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cauvi, D.M.; Hawisher, D.; Derunes, J.; De Maio, A. Phosphatidylcholine Liposomes Reprogram Macrophages toward an Inflammatory Phenotype. Membranes 2023, 13, 141. https://doi.org/10.3390/membranes13020141
Cauvi DM, Hawisher D, Derunes J, De Maio A. Phosphatidylcholine Liposomes Reprogram Macrophages toward an Inflammatory Phenotype. Membranes. 2023; 13(2):141. https://doi.org/10.3390/membranes13020141
Chicago/Turabian StyleCauvi, David M., Dennis Hawisher, Julia Derunes, and Antonio De Maio. 2023. "Phosphatidylcholine Liposomes Reprogram Macrophages toward an Inflammatory Phenotype" Membranes 13, no. 2: 141. https://doi.org/10.3390/membranes13020141
APA StyleCauvi, D. M., Hawisher, D., Derunes, J., & De Maio, A. (2023). Phosphatidylcholine Liposomes Reprogram Macrophages toward an Inflammatory Phenotype. Membranes, 13(2), 141. https://doi.org/10.3390/membranes13020141