
Ni–Cu High-Loaded Sol–Gel Catalysts for Dehydrogenation of Liquid Organic Hydrides: Insights into Structural Features and Relationship with Catalytic Activity

 , , , ,
, , , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Catalyst Preparation
2.3. Catalyst Testing in the Dehydrogenation of MCH
2.4. CO Chemisorption
2.5. Nitrogen Physisorption
2.6. Temperature-Programmed Reduction
2.7. X-ray Diffraction
2.8. High-Resolution Transmission Electron Microscopy
2.9. X-ray Photoelectron Spectroscopy
2.10. X-ray Absorption Spectroscopy
2.11. CHNS Analysis of Catalysts after Dehydrogenation of MCH
3. Results and Discussion
3.1. MCH Dehydrogenation
3.2. Catalyst Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brückner, N.; Obesser, K.; Bösmann, A.; Teichmann, D.; Arlt, W.; Dungs, J.; Wasserscheid, P. Evaluation of industrially applied heat-transfer fluids as liquid organic hydrogen carrier systems. ChemSusChem 2014, 7, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Aakko-Saksa, P.T.; Cook, C.; Kiviaho, J.; Repo, T. Liquid organic hydrogen carriers for transportation and storing of renewable energy—Review and discussion. J. Power Source 2018, 396, 803–823. [Google Scholar] [CrossRef]
- Brigljević, B.; Byun, M.; Lim, H. Design, economic evaluation, and market uncertainty analysis of LOHC-based, CO2 free, hydrogen delivery systems. Appl. Energy 2020, 274, 115314. [Google Scholar] [CrossRef]
- Crabtree, R.H. Hydrogen storage in liquid organic heterocycles. Energy Environ. Sci. 2008, 1, 134–138. [Google Scholar] [CrossRef]
- Moores, A.; Poyatos, M.; Luo, Y.; Crabtree, R.H. Catalysed low temperature H2 release from nitrogen heterocycles. New J. Chem. 2006, 30, 1675–1678. [Google Scholar] [CrossRef]
- Okada, Y.; Sasaki, E.; Watanabe, E.; Hyodo, S.; Nishijima, H. Development of dehydrogenation catalyst for hydrogen generation in organic chemical hydride method. Int. J. Hydrogen Energy 2006, 31, 1348–1356. [Google Scholar] [CrossRef]
- Niermann, M.; Beckendorff, A.; Kaltschmitt, M.; Bonhoff, K. Liquid organic hydrogen carrier (LOHC)—Assessment based on chemical and economic properties. Int. J. Hydrogen Energy 2019, 44, 6631–6654. [Google Scholar] [CrossRef]
- Biniwale, R.B.; Rayalu, S.; Devotta, S.; Ichikawa, M. Chemical hydrides: A solution to high capacity hydrogen storage and supply. Int. J. Hydrogen Energy 2008, 33, 360–365. [Google Scholar] [CrossRef]
- Alhumaidan, F.; Tsakiris, D.; Cresswell, D.; Garforth, A. Hydrogen storage in liquid organic hydride: Selectivity of MCH dehydrogenation over monometallic and bimetallic Pt catalysts. Int. J. Hydrogen Energy 2013, 38, 14010–14026. [Google Scholar] [CrossRef]
- Jothimurugesan, K.; Bhatia, S.; Srivastava, R.D. Kinetics of dehydrogenation of methylcyclohexane over a platinum-rhenium-alumina catalyst in the presence of added hydrogen. Ind. Eng. Chem. Fundam. 1985, 24, 433–438. [Google Scholar] [CrossRef]
- Foppa, L.; Dupont, J. Benzene partial hydrogenation: Advances and perspectives. Chem. Soc. Rev. 2015, 44, 1886–1897. [Google Scholar] [CrossRef]
- Menon, P.G.; Paál, Z. Some aspects of the mechanisms of catalytic reforming reactions. Ind. Eng. Chem. Res. 1997, 36, 3282–3291. [Google Scholar] [CrossRef]
- Sebastián, D.; Alegre, C.; Calvillo, L.; Pérez, M.; Moliner, R.; Lázaro, M.J. Carbon supports for the catalytic dehydrogenation of liquid organic hydrides as hydrogen storage and delivery system. Int. J. Hydrogen Energy 2014, 39, 4109–4115. [Google Scholar] [CrossRef] [Green Version]
- Biniwale, R.B.; Kariya, N.; Ichikawa, M. Dehydrogenation of cyclohexane over Ni based catalysts supported on activated carbon using spray-pulsed reactor and enhancement in activity by addition of a small amount of pt. Catal. Lett. 2005, 105, 83–87. [Google Scholar] [CrossRef]
- Preuster, P.; Papp, C.; Wasserscheid, P. Liquid organic hydrogen carriers (LOHCs): Toward a hydrogen-free hydrogen economy. Acc. Chem. Res. 2017, 50, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Shimura, M. Development of large scale H2 storage and transportation technology with liquid organic hydrogen carrier (LOHC). In Proceedings of the 21st Joint GCC-Japan Environment Symposium, Doha, Qatar, 5–6 February 2013. [Google Scholar]
- Gianotti, E.; Reyes-Carmona, Á.; Taillades-Jacquin, M.; Taillades, G.; Rozière, J.; Jones, D.J. Study of the effect of addition of In to Pt-Sn/γ-Al2O3 catalysts for high purity hydrogen production via partial dehydrogenation of kerosene jet A-1. Appl. Catal. B 2014, 160–161, 574–581. [Google Scholar] [CrossRef]
- Boufaden, N.; Akkari, R.; Pawelec, B.; Fierro, J.L.G.; Zina, M.S.; Ghorbel, A. Dehydrogenation of methylcyclohexane to toluene over partially reduced silica-supported Pt-Mo catalysts. J. Molec. Catal. A 2016, 420, 96–106. [Google Scholar] [CrossRef]
- Manabe, S.; Yabe, T.; Nakano, A.; Nagatake, S.; Higo, T.; Ogo, S.; Nakai, H.; Sekine, Y. Theoretical investigation on structural effects of Pt–Mn catalyst on activity and selectivity for methylcyclohexane dehydrogenation. Chem. Phys. Lett. 2018, 711, 73–76. [Google Scholar] [CrossRef]
- Yan, J.; Wang, W.; Miao, L.; Wu, K.; Chen, G.; Huang, Y.; Yang, Y. Dehydrogenation of methylcyclohexane over PtSn supported on MgAl mixed metal oxides derived from layered double hydroxides. Int. J. Hydrogen Energy 2018, 43, 9343–9352. [Google Scholar] [CrossRef]
- Rao, P.C.; Yoon, M. Potential liquid-organic hydrogen carrier (LOHC) systems: A review on recent progress. Energies 2020, 13, 6040. [Google Scholar] [CrossRef]
- Al-ShaikhAli, A.H.; Jedidi, A.; Cavallo, L.; Takanabe, K. Non-precious bimetallic catalysts for selective dehydrogenation of an organic chemical hydride system. Chem. Commun. 2015, 51, 12931–12934. [Google Scholar] [CrossRef] [Green Version]
- Belatel, H.; Al-Kandari, H.; Al-Khorafi, F.; Katrib, A.; Garin, F. Catalytic reactions of methylcyclohexane (MCH) on partially reduced MoO3. Appl. Catal. A 2004, 275, 141–147. [Google Scholar] [CrossRef]
- Boufaden, N.; Akkari, R.; Pawelec, B.; Fierro, J.L.G.; Said Zina, M.; Ghorbel, A. Dehydrogenation of methylcyclohexane to toluene over partially reduced Mo–SiO2 catalysts. Appl. Catal. A 2015, 502, 329–339. [Google Scholar] [CrossRef]
- Meng, J.; Zhou, F.; Ma, H.; Yuan, X.; Wang, Y.; Zhang, J. A review of catalysts for methylcyclohexane dehydrogenation. Top. Catal. 2021, 64, 509–520. [Google Scholar] [CrossRef]
- Desai, P.H.; Richardson, J.T. Crystallite size effects in nickel catalysts: Cyclohexane dehydrogenation and hydrogenolysis. J. Catal. 1986, 98, 392–400. [Google Scholar] [CrossRef]
- Xia, Z.; Lu, H.; Liu, H.; Zhang, Z.; Chen, Y. Cyclohexane dehydrogenation over Ni-Cu/SiO2 catalyst: Effect of copper addition. Catal. Commun. 2017, 90, 39–42. [Google Scholar] [CrossRef]
- Ermakov, D.Y.; Bykova, M.V.; Selisheva, S.A.; Khromova, S.A.; Yakovlev, V.A. Method of Preparing Hydrotreatment Catalyst. Patent RU249 6580C1, 2013. [Google Scholar]
- Ermakova, M.A.; Ermakov, D.Y. High-loaded nickel–silica catalysts for hydrogenation, prepared by sol–gel. Route: Structure and catalytic behavior. Appl. Catal. A 2003, 245, 277–288. [Google Scholar] [CrossRef]
- Tsybulya, S.V.; Cherepanova, S.V.; Soloviyova, L.P. Polycrystal software package for IBM/PC. J. Struct. Chem. 1996, 37, 332–334. [Google Scholar] [CrossRef]
- Kaichev, V.V.; Chesalov, Y.A.; Saraev, A.A.; Tsapina, A.M. A mechanistic study of dehydrogenation of propane over vanadia–titania catalysts. J. Phys. Chem. C 2019, 123, 19668–19680. [Google Scholar] [CrossRef]
- Fairley, N. CasaXPS: Processing Software for XPS, AES, SIMS and More. Available online: http://www.casaxps.com (accessed on 14 July 2021).
- Scofield, J.H. Hartree-slater subshell photoionization cross-sections at 1254 and 1487 ev. J. Electron Spectrosc. Relat. Phenom. 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Chernyshov, A.A.; Veligzhanin, A.A.; Zubavichus, Y.V. Structural materials science end-station at the Kurchatov synchrotron radiation source: Recent instrumentation upgrades and experimental results. Nucl. Instrum. Methods Phys. Res. Sect. A 2009, 603, 95–98. [Google Scholar] [CrossRef]
- Rehr, J.J.; Mustre de Leon, J.; Zabinsky, S.I.; Albers, R.C. Theoretical X-ray absorption fine structure standards. J. Am. Chem. Soc. 1991, 113, 5135–5140. [Google Scholar] [CrossRef]
- Gulyaeva, Y.K.; Alekseeva, M.V.; Ermakov, D.Y.; Bulavchenko, O.A.; Zaikina, O.O.; Yakovlev, V.A. High-loaded nickel based sol–gel catalysts for methylcyclohexane dehydrogenation. Catalysts 2020, 10, 1198. [Google Scholar] [CrossRef]
- Escobar, J.; De Los Reyes, J.A.; Viveros, T.; Barrera, M.C. Cyclohexane dehydrogenation over wet-impregnated Ni on Al2O3−TiO2 sol−gel oxides. Ind. Eng. Chem. Res. 2006, 45, 5693–5700. [Google Scholar] [CrossRef]
- Zhang, X.; He, N.; Lin, L.; Zhu, Q.; Wang, G.; Guo, H. Study of the carbon cycle of a hydrogen supply system over a supported Pt catalyst: Methylcyclohexane–toluene–hydrogen cycle. Catal. Sci. Technol. 2020, 10, 1171–1181. [Google Scholar] [CrossRef]
- Patil, S.P.; Pande, J.V.; Biniwale, R.B. Non-noble Ni–Cu/ACC bimetallic catalyst for dehydrogenation of liquid organic hydrides for hydrogen storage. Int. J. Hydrogen Energy 2013, 38, 15233–15241. [Google Scholar] [CrossRef]
- Alekseeva, M.V.; Otyuskaya, D.S.; Rekhtina, M.A.; Bulavchenko, O.A.; Stonkus, O.A.; Kaichev, V.V.; Zavarukhin, S.G.; Thybaut, J.W.; Alexiadis, V.; Venderbosch, R.H.; et al. NiCuMo-SiO2 catalyst for pyrolysis oil upgrading: Model acidic treatment study. Appl. Catal. A 2019, 573, 1–12. [Google Scholar] [CrossRef]
- Asedegbega-Nieto, E.; Guerrero-Ruíz, A.; Rodríguez-Ramos, I. Study of co chemisorption on graphite-supported Ru–Cu and Ni–Cu bimetallic catalysts. Thermochim. Acta 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Pannell, R.B. The stoichiometry of hydrogen and carbon monoxide chemisorption on alumina- and silica-supported nickel. J. Catal. 1980, 65, 390–401. [Google Scholar] [CrossRef]
- Ueckert, T.; Lamber, R.; Jaeger, N.I.; Schubert, U. Strong metal support interactions in a Ni/SiO2 catalyst prepared via sol-gel synthesis. Appl. Catal. A 1997, 155, 75–85. [Google Scholar] [CrossRef]
- Crisafulli, C.; Galvagno, S.; Maggiore, R.; Scirè, S.; Saeli, A. Performance of supported Ru-Cu bimetallic catalysts prepared from nitrate precursors. Catal. Lett. 1990, 6, 77–83. [Google Scholar] [CrossRef]
- Thyssen, V.V.; Maia, T.A.; Assaf, E.M. Cu and Ni catalysts supported on γ-Al2O3 and SiO2 assessed in glycerol steam reforming reaction. J. Braz. Chem. Soc. 2015, 26, 22–31. [Google Scholar]
- Srivastava, S.; Jadeja, G.C.; Parikh, J. Synergism studies on alumina-supported copper-nickel catalysts towards furfural and 5-hydroxymethylfurfural hydrogenation. J. Mol. Catal. A 2017, 426, 244–256. [Google Scholar] [CrossRef]
- Fedorov, A.V.; Kukushkin, R.G.; Yeletsky, P.M.; Bulavchenko, O.A.; Chesalov, Y.A.; Yakovlev, V.A. Temperature-programmed reduction of model CuO, NiO and mixed CuO–NiO catalysts with hydrogen. J. Alloys Compd. 2020, 844, 156135. [Google Scholar] [CrossRef]
- Ardiyanti, A.R.; Bykova, M.V.; Khromova, S.A.; Yin, W.; Venderbosch, R.H.; Yakovlev, V.A.; Heeres, H.J. Ni-based catalysts for the hydrotreatment of fast pyrolysis oil. Energy Fuels 2016, 30, 1544–1554. [Google Scholar] [CrossRef]
- Robertson, S.D.; McNicol, B.D.; De Baas, J.H.; Kloet, S.C.; Jenkins, J.W. Determination of reducibility and identification of alloying in copper-nickel-on-silica catalysts by temperature-programmed reduction. J. Catal. 1975, 37, 424–431. [Google Scholar] [CrossRef]
- Naghash, A.R.; Etsell, T.H.; Xu, S. XRD and XPS study of Cu−Ni interactions on reduced copper−nickel−aluminum oxide solid solution catalysts. Chem. Mater. 2006, 18, 2480–2488. [Google Scholar] [CrossRef]
- Gutowski, M.; Jaffe, J.E.; Liu, C.-L.; Stoker, M.; Hegde, R.I.; Rai, R.S.; Tobin, P.J. Thermodynamic stability of high-K dielectric metal oxides ZrO2 and HfO2 in contact with Si and SiO2. Appl. Phys. Lett. 2002, 80, 1897–1899. [Google Scholar] [CrossRef]
- Kim, S.; Kim, M.C.; Choi, S.-H.; Kim, K.J.; Hwang, H.N.; Hwang, C.C. Size dependence of Si 2p core-level shift at Si nanocrystal/SiO2 interfaces. Appl. Phys. Lett. 2007, 91, 103113. [Google Scholar] [CrossRef]
- Mendialdua, J.; Casanova, R.; Rueda, F.; Rodríguez, A.; Quiñones, J.; Alarcón, L.; Escalante, E.; Hoffmann, P.; Taebi, I.; Jalowiecki, L. X-ray photoelectron spectroscopy studies of laterite standard reference material. J. Mol. Catal. A 2005, 228, 151–162. [Google Scholar] [CrossRef]
- Occelli, M.L.; Psaras, D.; Suib, S.L.; Stencel, J.M. Metal contaminant effects on the properties of a silica-rich fluid cracking catalyst. Appl. Catal. 1986, 28, 143–160. [Google Scholar] [CrossRef]
- Shalvoy, R.B.; Reucroft, P.J.; Davis, B.H. Characterization of coprecipitated nickel on silica methanation catalysts by X-ray photoelectron spectroscopy. J. Catal. 1979, 56, 336–348. [Google Scholar] [CrossRef]
- Kaichev, V.V.; Teschner, D.; Saraev, A.A.; Kosolobov, S.S.; Gladky, A.Y.; Prosvirin, I.P.; Rudina, N.A.; Ayupov, A.B.; Blume, R.; Hävecker, M.; et al. Evolution of self-sustained kinetic oscillations in the catalytic oxidation of propane over a nickel foil. J. Catal. 2016, 334, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Khromova, S.A.; Smirnov, A.A.; Bulavchenko, O.A.; Saraev, A.A.; Kaichev, V.V.; Reshetnikov, S.I.; Yakovlev, V.A. Anisole hydrodeoxygenation over Ni–Cu bimetallic catalysts: The effect of Ni/Cu ratio on selectivity. Appl. Catal. A 2014, 470, 261–270. [Google Scholar] [CrossRef]
- Fedorov, A.; Saraev, A.; Kremneva, A.; Selivanova, A.; Vorokhta, M.; Šmíd, B.; Bulavchenko, O.; Yakovlev, V.; Kaichev, V. Kinetic and mechanistic study of co oxidation over nanocomposite Cu−Fe−Al oxide catalysts. ChemCatChem 2020, 12, 4911–4921. [Google Scholar] [CrossRef]
- Richter, M.; Fait, M.J.G.; Eckelt, R.; Schneider, M.; Radnik, J.; Heidemann, D.; Fricke, R. Gas-phase carbonylation of methanol to dimethyl carbonate on chloride-free Cu-precipitated zeolite Y at normal pressure. J. Catal. 2007, 245, 11–24. [Google Scholar] [CrossRef]
- Bykova, M.V.; Ermakov, D.Y.; Khromova, S.A.; Smirnov, A.A.; Lebedev, M.Y.; Yakovlev, V.A. Stabilized Ni-based catalysts for bio-oil hydrotreatment: Reactivity studies using guaiacol. Catal. Today 2014, 220–222, 21–31. [Google Scholar] [CrossRef]
- West, A.R. Solid State Chemistry and Its Applications, 2nd ed.; student ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2014; p. 556. [Google Scholar]
- Sinfelt, J.H.; Carter, J.L.; Yates, D.J.C. Catalytic hydrogenolysis and dehydrogenation over copper-nickel alloys. J. Catal. 1972, 24, 283–296. [Google Scholar] [CrossRef]
- Gandarias, I.; Requies, J.; Arias, P.L.; Armbruster, U.; Martin, A. Liquid-phase glycerol hydrogenolysis by formic acid over Ni–Cu/Al2O3 catalysts. J. Catal. 2012, 290, 79–89. [Google Scholar] [CrossRef]
- Coq, B.; Figueras, F. Bimetallic palladium catalysts: Influence of the co-metal on the catalyst performance. J. Mol. Catal. A 2001, 173, 117–134. [Google Scholar] [CrossRef]
- Bearden, J.A.; Burr, A.F. Reevaluation of X-ray atomic energy levels. Rev. Mod. Phys. 1967, 39, 125–142. [Google Scholar] [CrossRef]
- Rochet, A.; Baubet, B.; Moizan, V.; Devers, E.; Hugon, A.; Pichon, C.; Payen, E.; Briois, V. Influence of the preparation conditions of oxidic NiMo/Al2O3 catalysts on the sulfidation ability: A quick-XAS and RAMAN spectroscopic study. J. Phys. Chem. C 2015, 119, 23928–23942. [Google Scholar] [CrossRef]
- Preda, I.; Soriano, L.; Díaz-Fernández, D.; Domínguez-Cañizares, G.; Gutiérrez, A.; Castro, G.R.; Chaboy, J. X-ray absorption study of the local structure at the NiO/oxide interfaces. J. Synchrotron Radiat. 2013, 20, 635–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
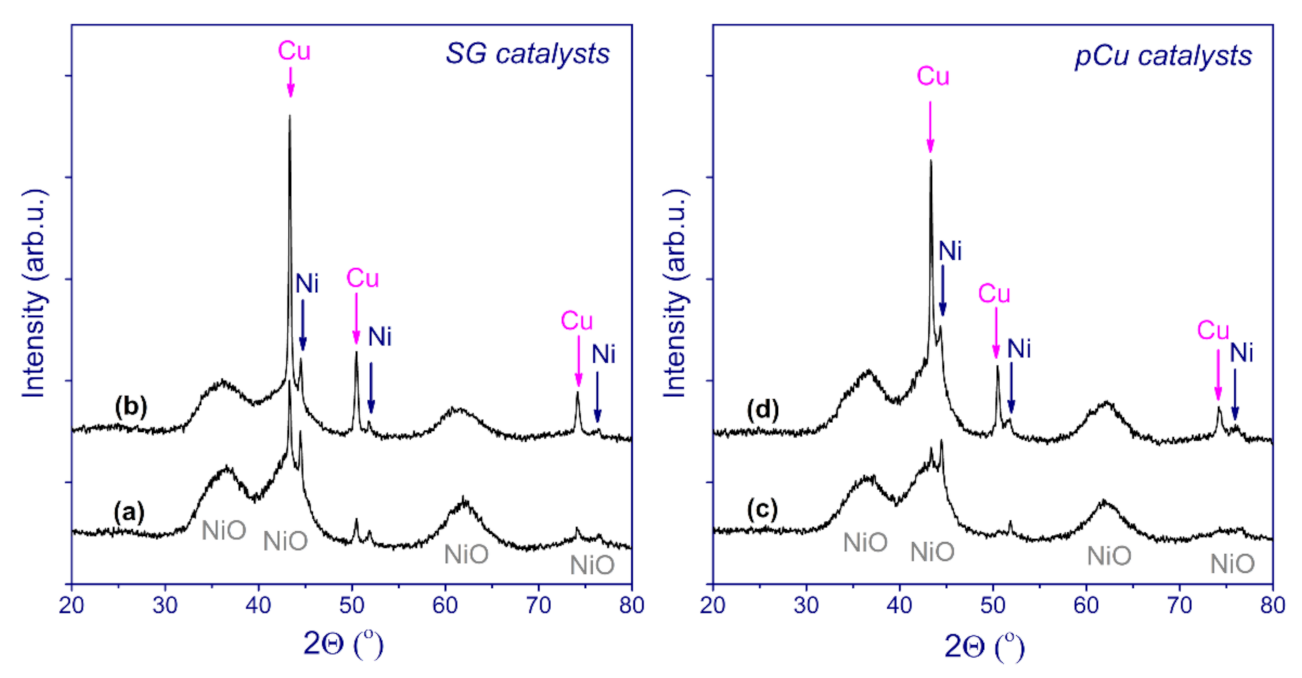{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst a | Composition d, wt% | Atomic Cu/Ni Ratio | Atomic Si/Ni Ratio | SSA e, m2/gcat | ||
|---|---|---|---|---|---|---|
| Cu | Ni | Si | ||||
| SG series b | ||||||
| Ni–SiO2 | - | 62.3 | 9.7 | - | 0.33 | 294 |
| Cu5Ni95–SiO2 | 3.0 | 59.3 | 9.7 | 0.05 | 0.34 | 163 |
| Cu10Ni90–SiO2 | 6.3 | 56.1 | 9.7 | 0.1 | 0.36 | 293 |
| Cu20Ni80–SiO2 | 12.5 | 49.9 | 9.7 | 0.23 | 0.41 | 259 |
| pCu series c | ||||||
| Cu5/Ni95–SiO2 | 3.3 | 59.9 | 9.3 | 0.05 | 0.33 | 234 |
| Cu10/Ni90–SiO2 | 6.4 | 57.4 | 8.9 | 0.1 | 0.33 | 262 |
| Cu20/Ni80–SiO2 | 13.0 | 52.2 | 8.1 | 0.23 | 0.33 | 235 |
| Cu30/Ni70–SiO2 | 19.9 | 46.7 | 7.3 | 0.4 | 0.33 | 157 |
| Catalyst | MCH Conversion, mol.% | Selectivity to TOL, mol.% | Yield of TOL, mol.% |
|---|---|---|---|
| SG series | |||
| Ni–SiO2 | 81 | 40 | 31 |
| Cu5Ni95–SiO2 | 76 | 49 | 37 |
| Cu10Ni90–SiO2 | 81 | 52 | 42 |
| Cu20Ni80–SiO2 | 87 | 56 | 49 |
| pCu series | |||
| Cu5/Ni95–SiO2 | 75 | 53 | 40 |
| Cu10/Ni90–SiO2 | 88 | 57 | 50 |
| Cu20/Ni80–SiO2 | 83 | 70 | 58 |
| Cu30/Ni70–SiO2 | 78 | 77 | 60 |
| Catalyst | CO Uptake, μmol/gcat |
|---|---|
| SG series | |
| Ni–SiO2 | 555 |
| Cu5Ni95–SiO2 | 449 |
| Cu10Ni90–SiO2 | 421 |
| Cu20Ni80–SiO2 | 385 |
| pCu series | |
| Cu5/Ni95–SiO2 | 384 |
| Cu10/Ni90–SiO2 | 358 |
| Cu20/Ni80–SiO2 | 266 |
| Cu30/Ni70–SiO2 | 213 |
| Catalyst | Ni/Si | Cu/Si | Cu/Ni |
|---|---|---|---|
| Cu20/Ni80–SiO2 | 0.45 | 0.11 | 0.244 |
| Cu5/Ni95–SiO2 | 0.56 | 0.04 | 0.071 |
| Cu20Ni80–SiO2 | 0.62 | 0.06 | 0.097 |
| Cu5Ni95–SiO2 | 0.61 | 0.01 | 0.016 |
| Catalyst | Lattice Parameter of Ni, Å | Cu/(Cu + Ni) Atomic (XRF) | NixCu1-x Composition (XRD) |
|---|---|---|---|
| Cu5Ni95–SiO2 | 3.526 | 0.046 | Ni0.95Cu0.05 |
| Cu20Ni80–SiO2 | 3.525 | 0.188 | Ni0.96Cu0.04 |
| Cu5/Ni95–SiO2 | 3.528 | 0.048 | Ni0.93Cu0.07 |
| Cu20/Ni80–SiO2 | 3.540 | 0.187 | Ni0.81 Cu0.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gulyaeva, Y.; Alekseeva, M.; Bulavchenko, O.; Kremneva, A.; Saraev, A.; Gerasimov, E.; Selishcheva, S.; Kaichev, V.; Yakovlev, V. Ni–Cu High-Loaded Sol–Gel Catalysts for Dehydrogenation of Liquid Organic Hydrides: Insights into Structural Features and Relationship with Catalytic Activity. Nanomaterials 2021, 11, 2017. https://doi.org/10.3390/nano11082017
Gulyaeva Y, Alekseeva M, Bulavchenko O, Kremneva A, Saraev A, Gerasimov E, Selishcheva S, Kaichev V, Yakovlev V. Ni–Cu High-Loaded Sol–Gel Catalysts for Dehydrogenation of Liquid Organic Hydrides: Insights into Structural Features and Relationship with Catalytic Activity. Nanomaterials. 2021; 11(8):2017. https://doi.org/10.3390/nano11082017
Chicago/Turabian StyleGulyaeva, Yuliya, Maria Alekseeva (Bykova), Olga Bulavchenko, Anna Kremneva, Andrey Saraev, Evgeny Gerasimov, Svetlana Selishcheva, Vasily Kaichev, and Vadim Yakovlev. 2021. "Ni–Cu High-Loaded Sol–Gel Catalysts for Dehydrogenation of Liquid Organic Hydrides: Insights into Structural Features and Relationship with Catalytic Activity" Nanomaterials 11, no. 8: 2017. https://doi.org/10.3390/nano11082017
APA StyleGulyaeva, Y., Alekseeva, M., Bulavchenko, O., Kremneva, A., Saraev, A., Gerasimov, E., Selishcheva, S., Kaichev, V., & Yakovlev, V. (2021). Ni–Cu High-Loaded Sol–Gel Catalysts for Dehydrogenation of Liquid Organic Hydrides: Insights into Structural Features and Relationship with Catalytic Activity. Nanomaterials, 11(8), 2017. https://doi.org/10.3390/nano11082017