
Hybrid Biochar/Ceria Nanomaterials: Synthesis, Characterization and Activity Assessment for the Persulfate-Induced Degradation of Antibiotic Sulfamethoxazole





Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
2.3. Physicochemical Characterization
2.4. Catalytic Activity
3. Results and Discussion
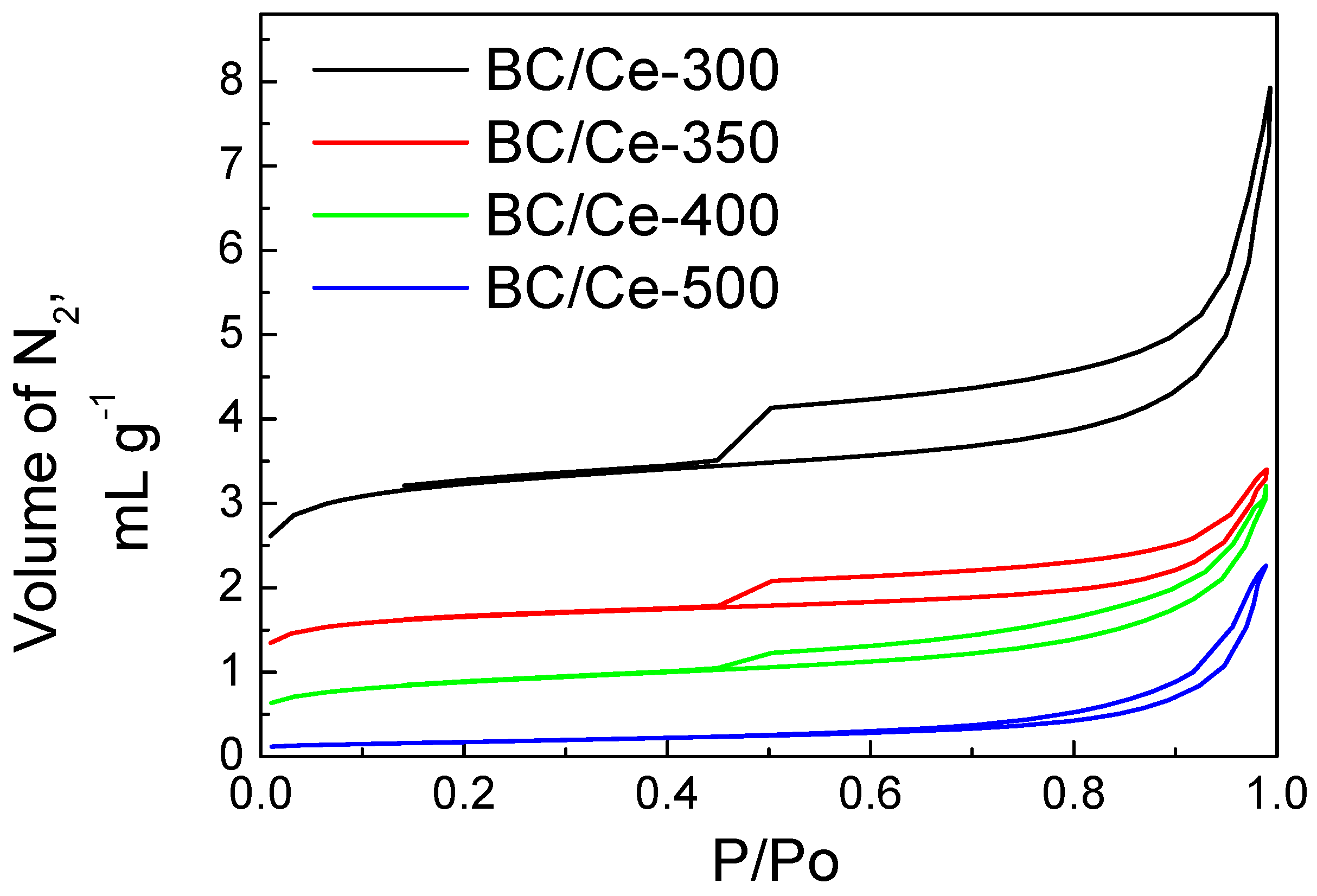
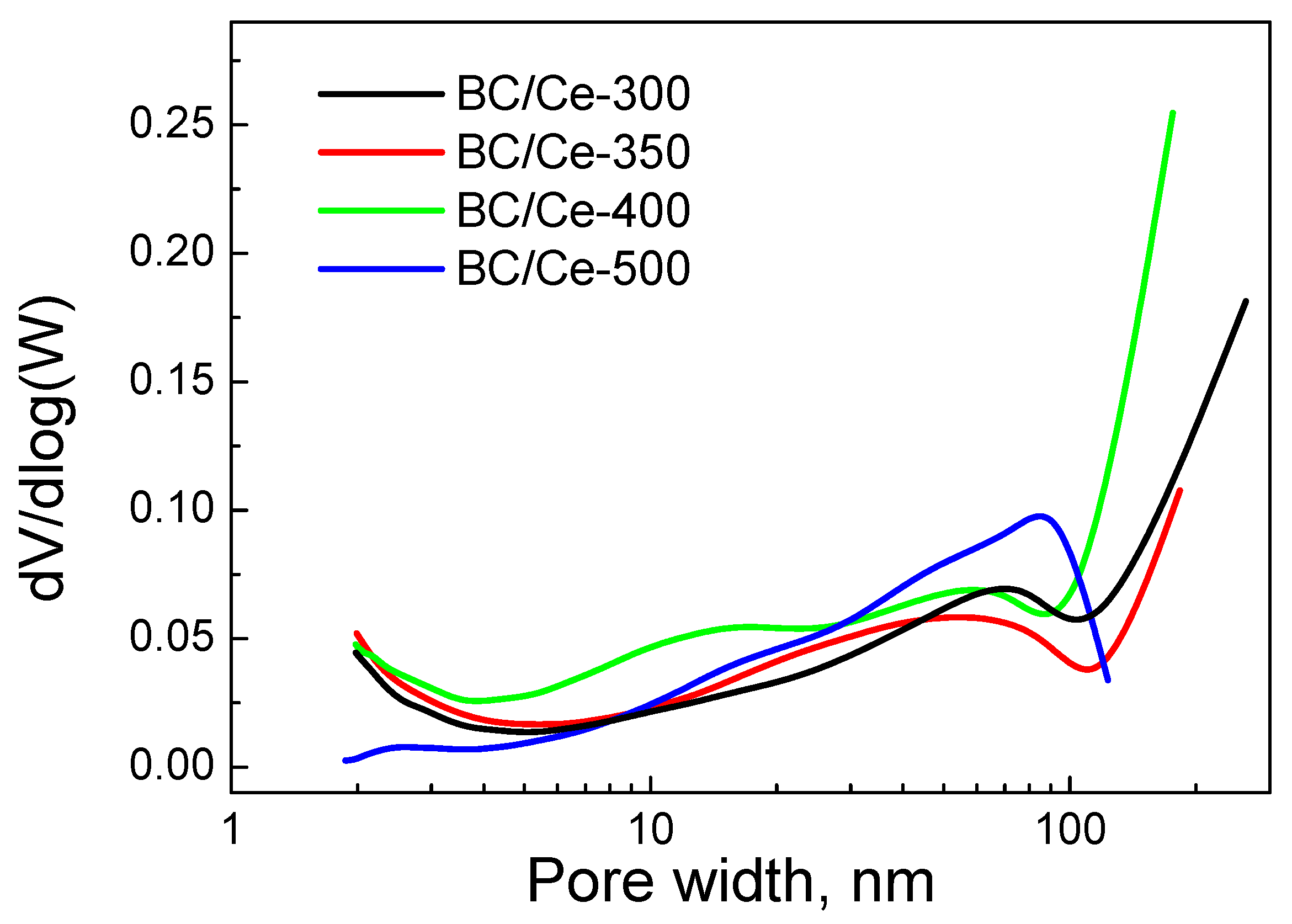
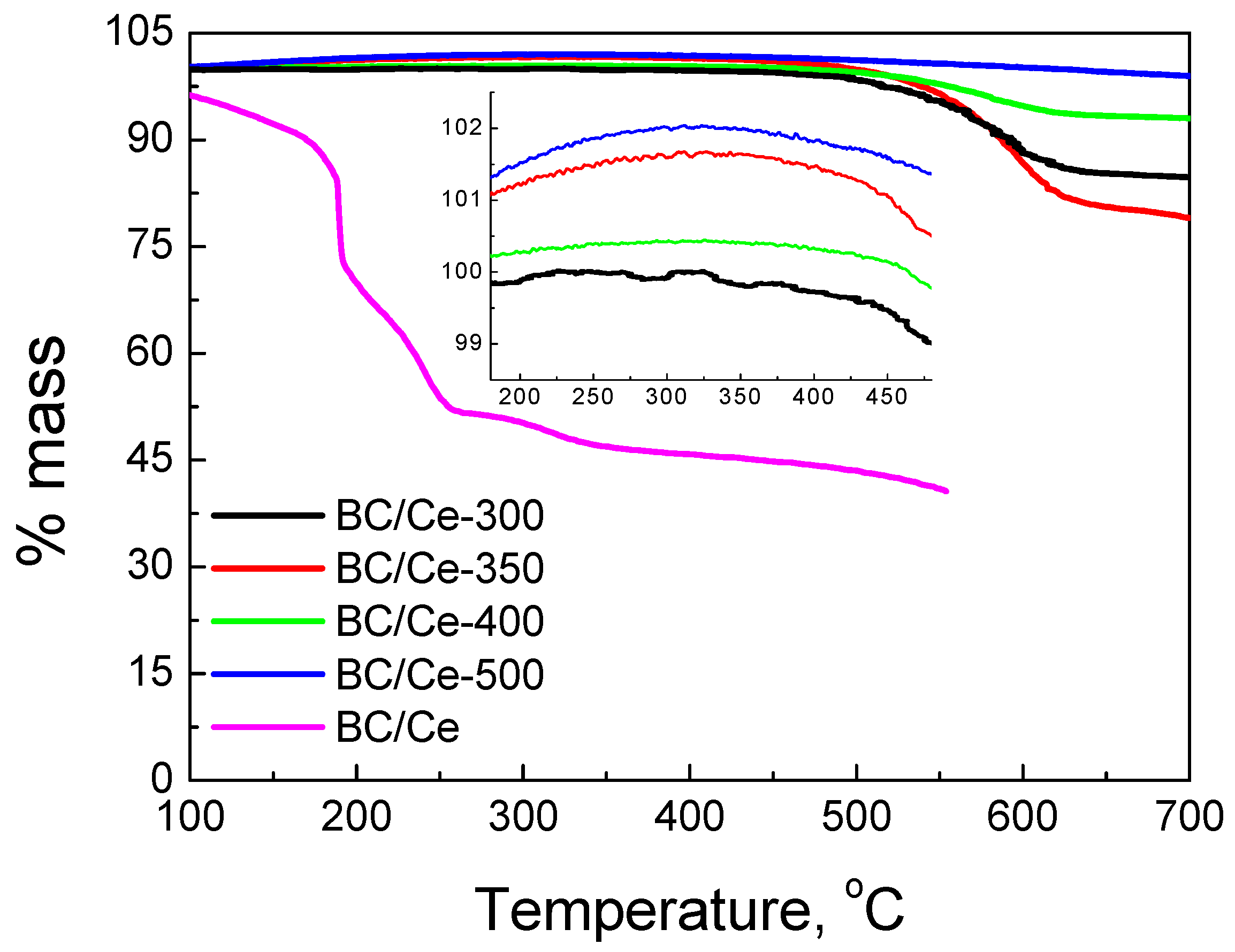
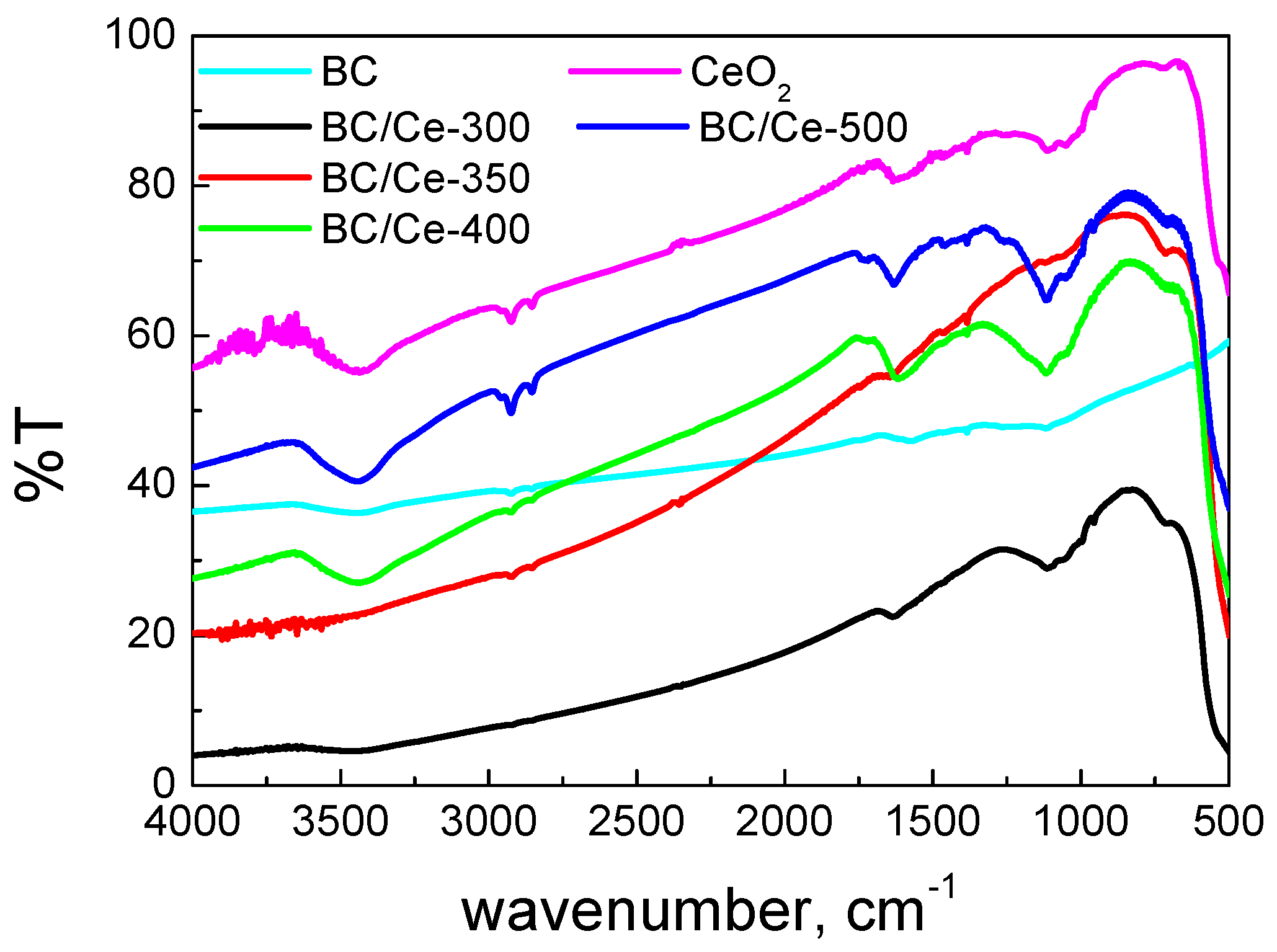
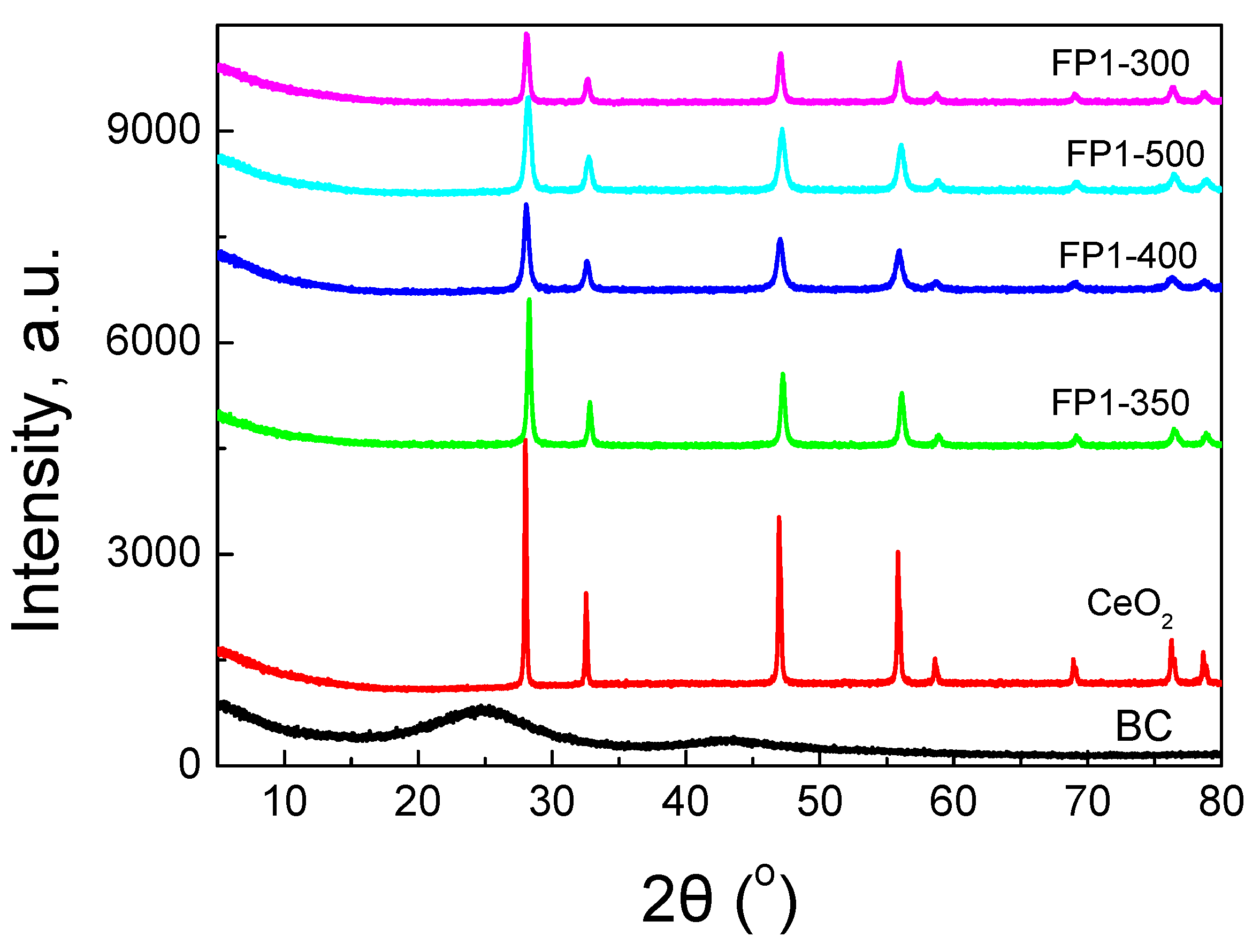
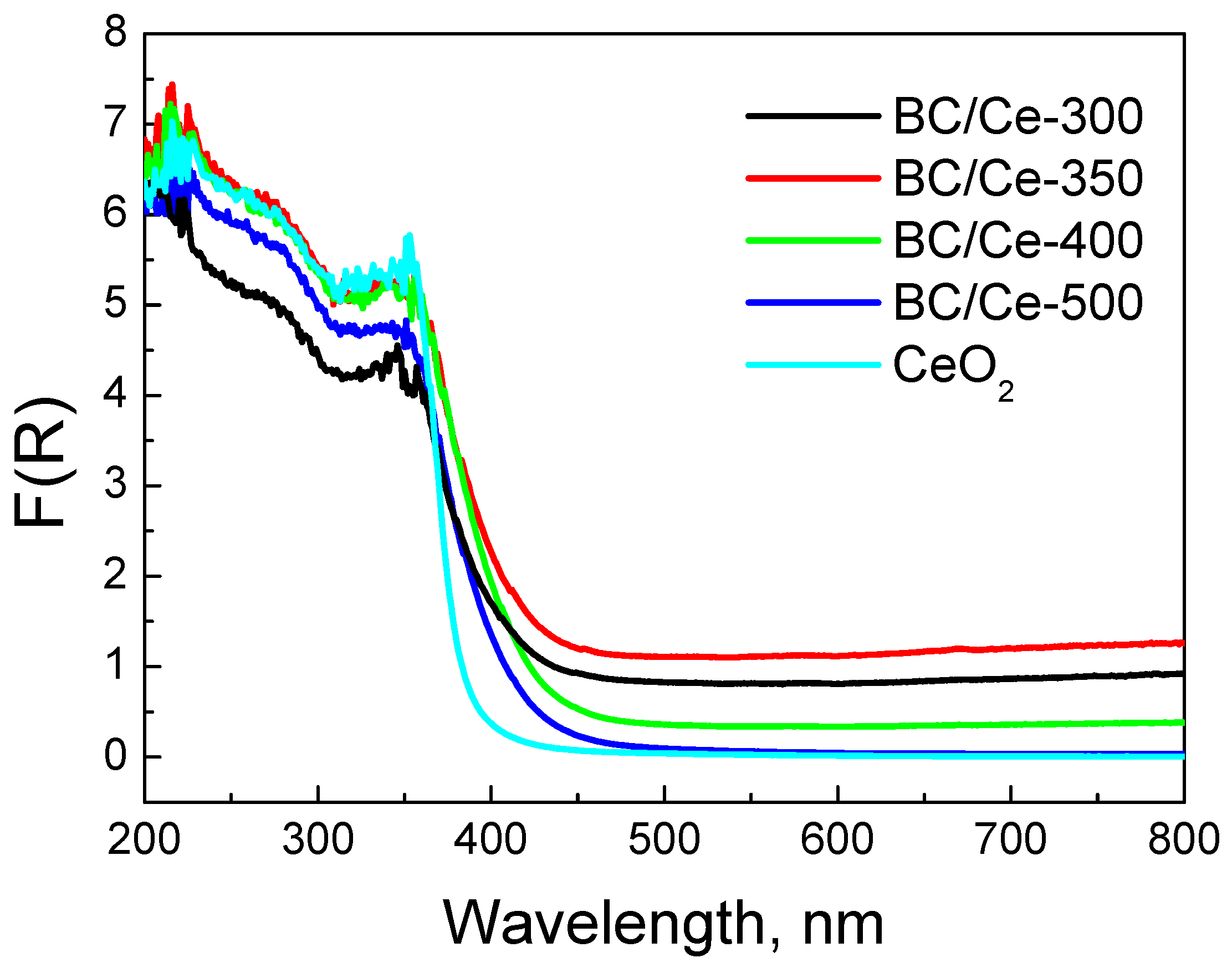
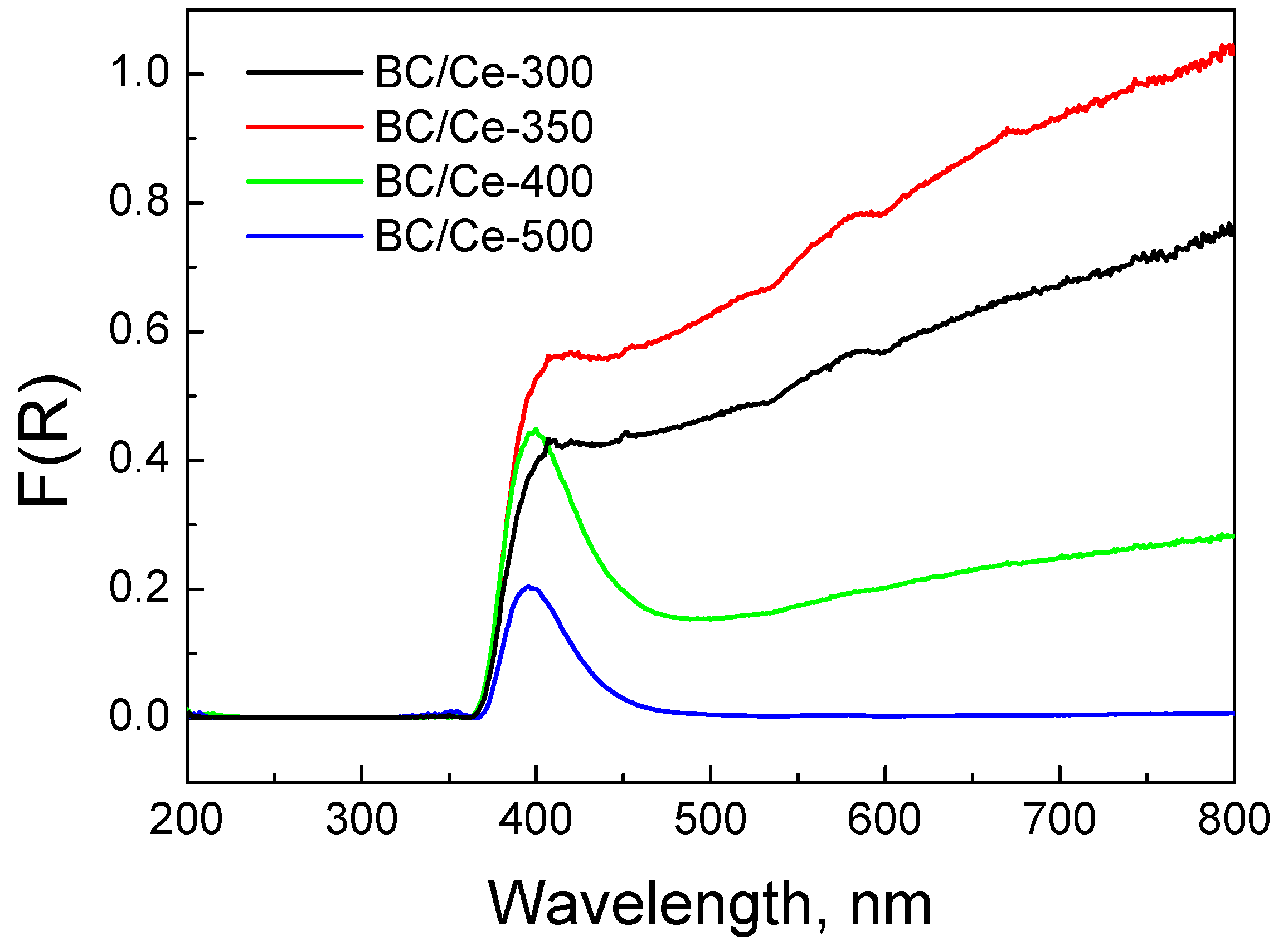
3.1. Samples Characterization
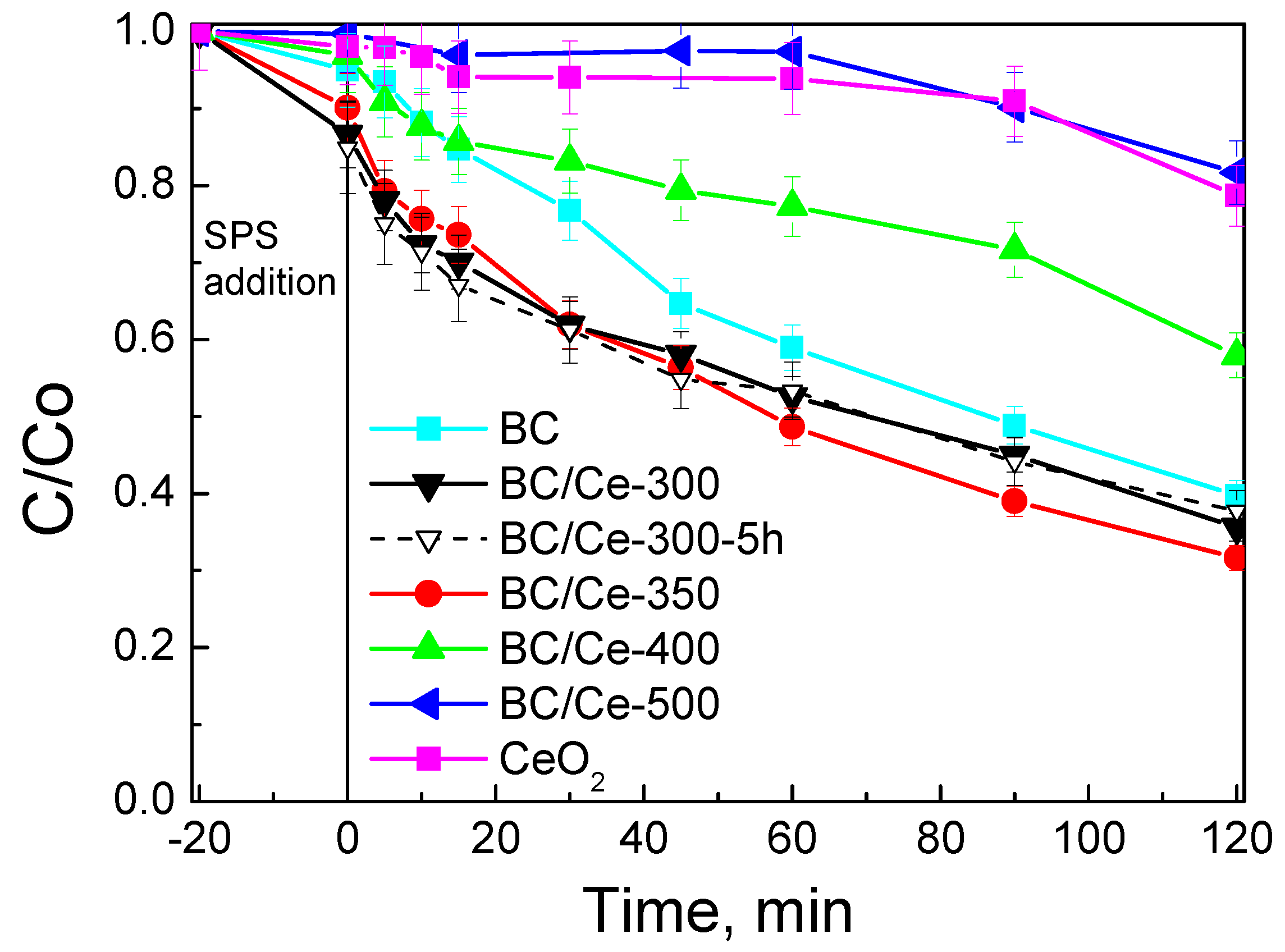
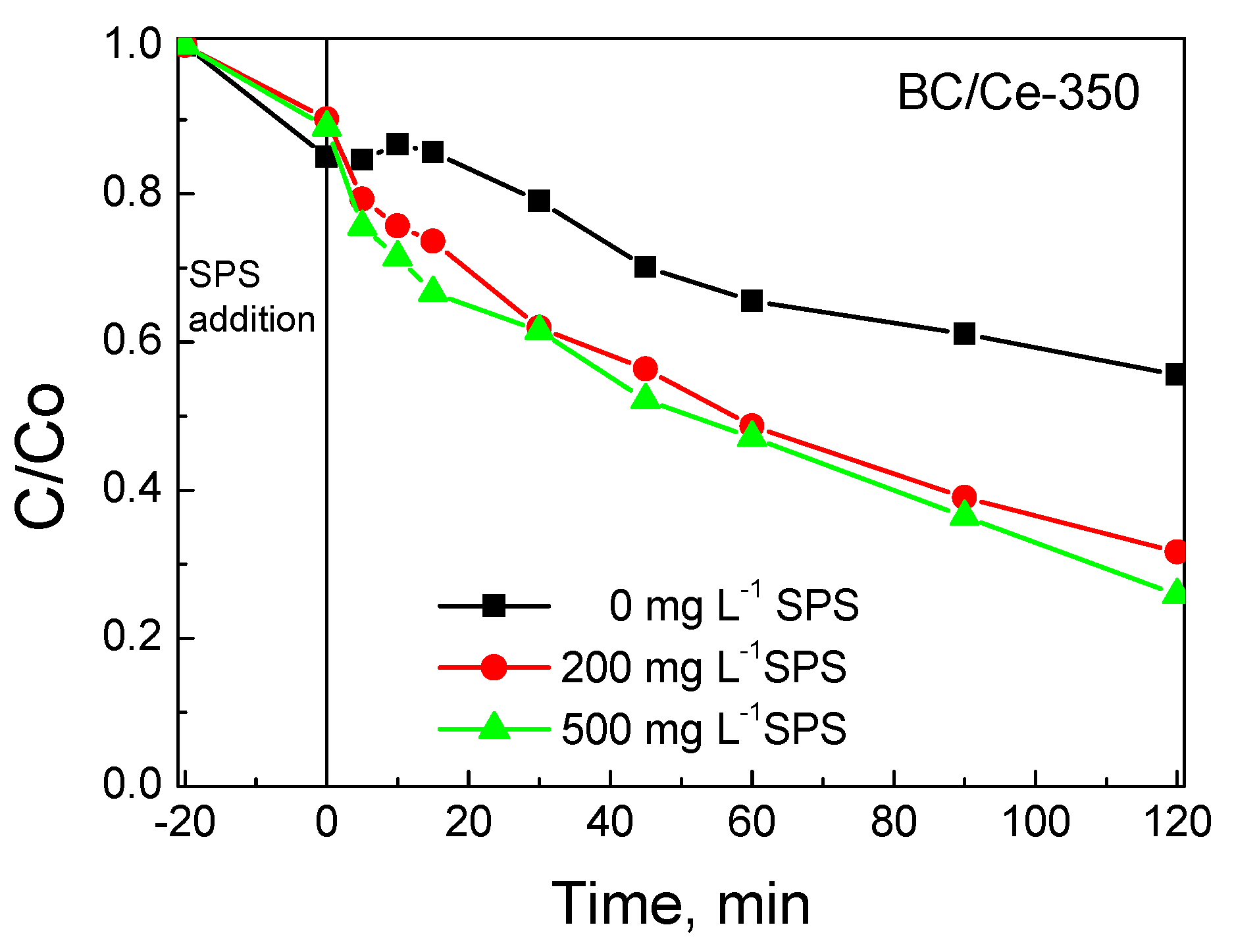
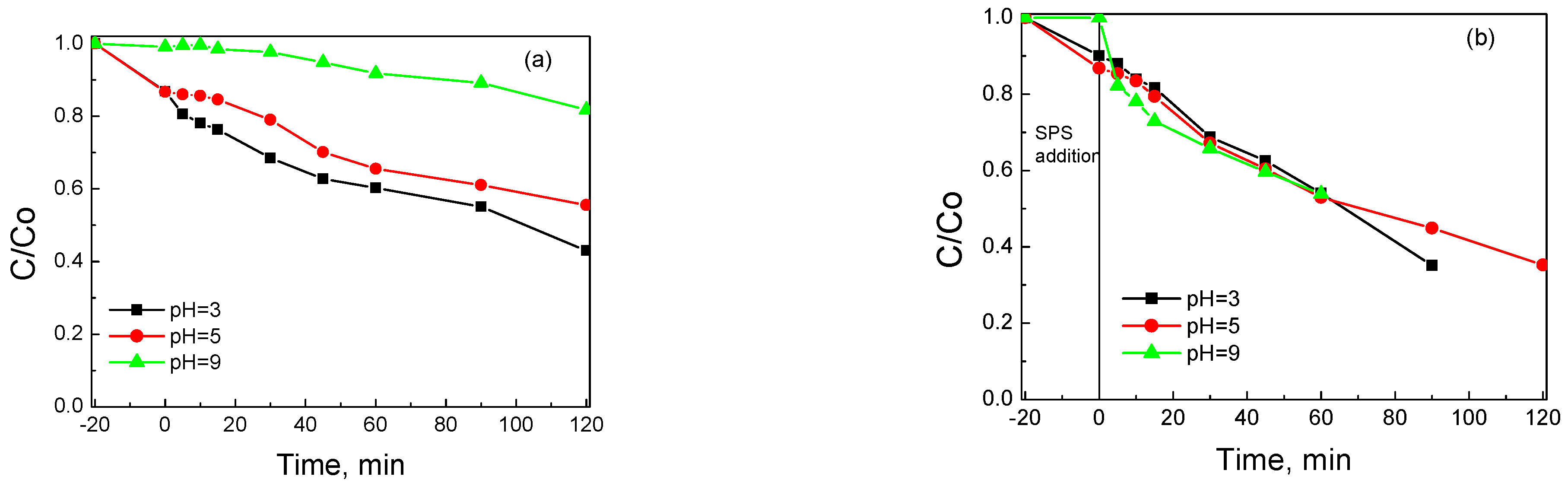
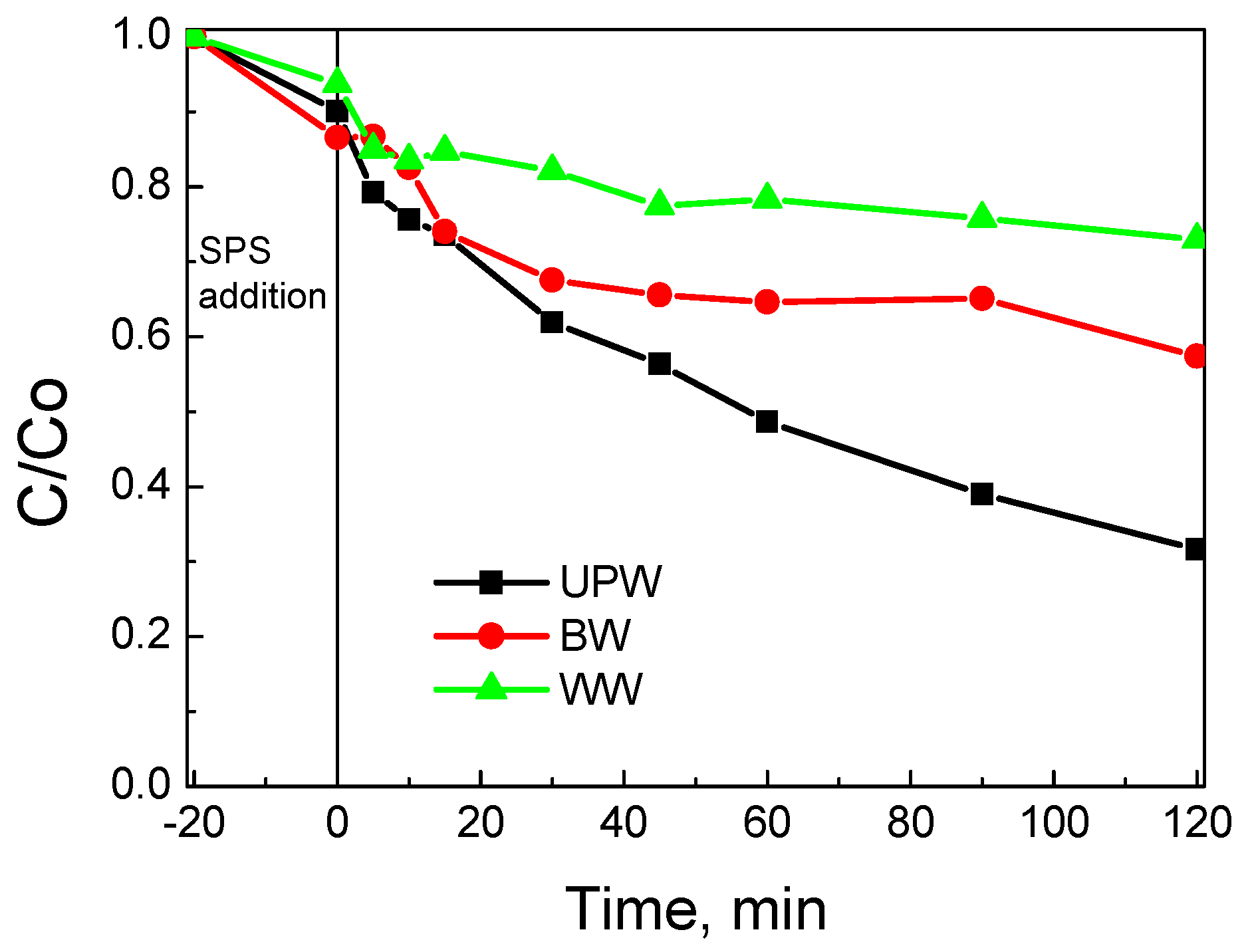
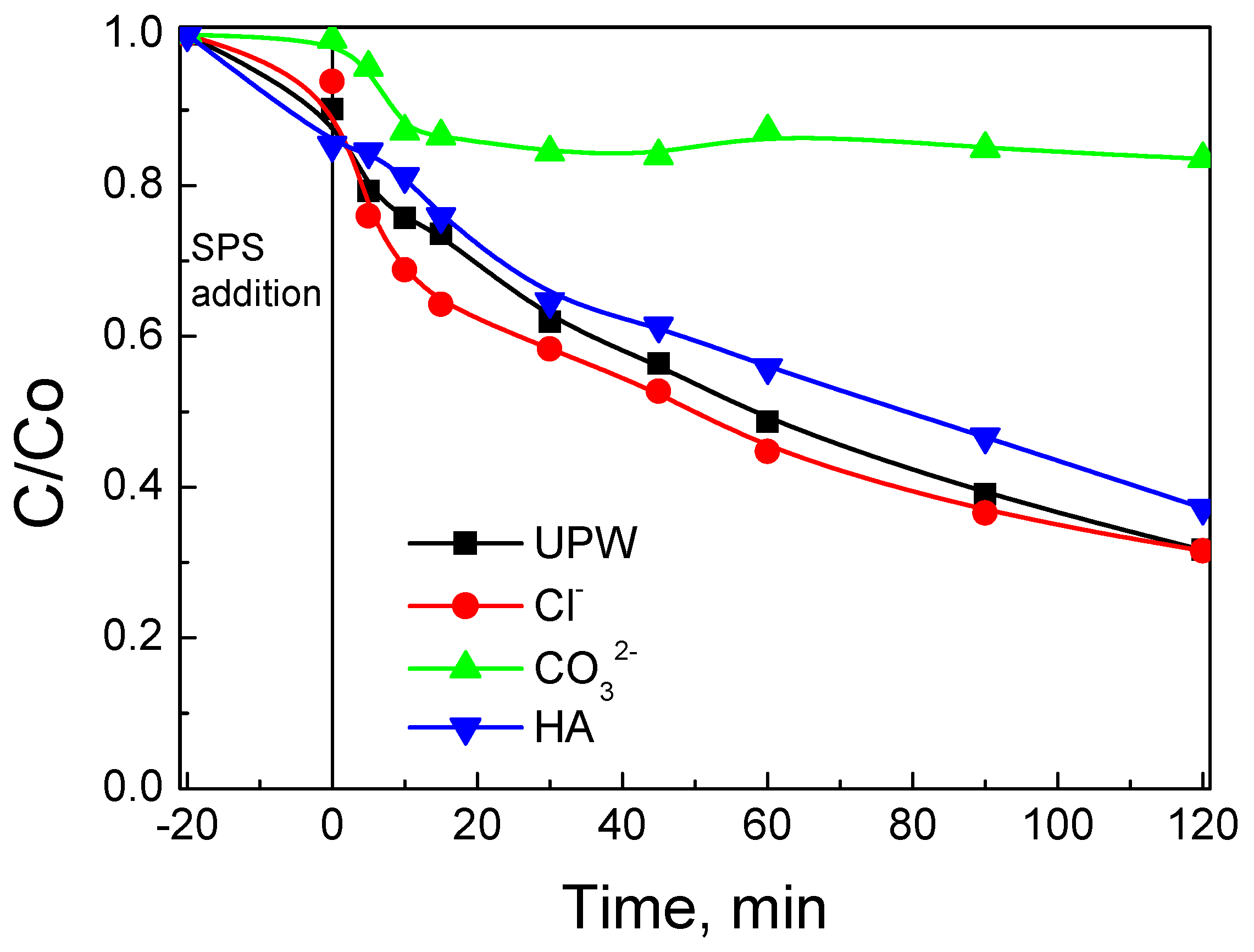
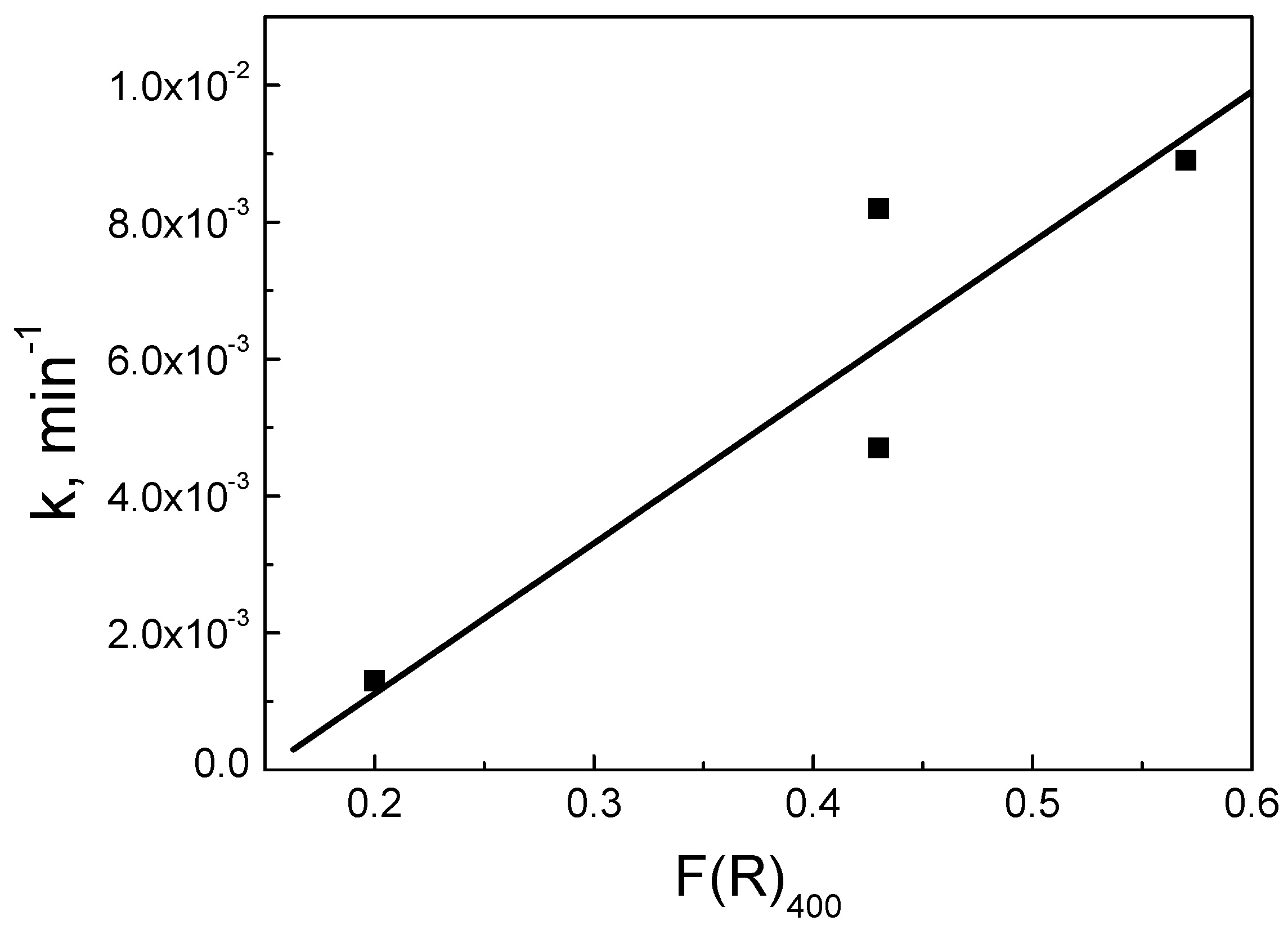
3.2. Assessment of Catalytic Activity for the Degradation of SMX
4. Conclusions
- Changing the calcination temperature in the range of –500 °C affected the biochar content and the physicochemical properties of CeO2, but more importantly, determines the interactions between biochar and CeO2 and, eventually, the catalytic activity.
- Calcination at 300–350 °C yielded the more active materials for persulfate activation and sulfamethoxazole degradation; the latter following pseudo-first order kinetics with the rate depending on the operating conditions.
- The water matrix is crucial for process performance since various inorganic and/or organic species can interfere with the surface and/or the target contaminant for the oxidants and the active catalytic sites. Hybrid materials may minimize such competitive interactions that do not exist in model experiments performed in pure water. Should this be the case, hybrid materials are likely to outperform bare biochar in environmentally relevant systems.
- Radicals and singlet oxygen seem to be the main oxidative species, as indirectly evidenced by means of scavenging experiments.
Author Contributions
Funding
Conflicts of Interest
References
- Fauzi, A.A.; Jalil, A.A.; Hassan, N.S.; Aziz, F.F.A.; Azami, M.S.; Hussain, I.; Saravanan, R.; Vo, D.-V.N. A critical review on relationship of CeO2-based photocatalyst towards mechanistic degradation of organic pollutant. Chemosphere 2022, 286, 131651. [Google Scholar] [CrossRef]
- Kappis, K.; Papadopoulos, C.; Papavasiliou, J.; Vakros, J.; Georgiou, Y.; Deligiannakis, Y.; Avgouropoulos, G. Tuning the Catalytic Properties of Copper-Promoted Nanoceria via a Hydrothermal Method. Catalysts 2019, 9, 138. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Singh, E.; Mishra, R.; Kumar, S. Biochar as environmental armour and its diverse role towards protecting soil, water and air. Sci. Total Environ. 2022, 806, 150444. [Google Scholar] [CrossRef]
- Ntzoufra, P.; Vakros, J.; Frontistis, Z.; Tsatsos, S.; Kyriakou, G.; Kennou, S.; Manariotis, I.D.; Mantzavinos, D. Effect of Sodium Persulfate Treatment on the Physicochemical Properties and Catalytic Activity of Biochar Prepared from Spent Malt Rootlets. J. Environ. Chem. Eng. 2021, 9, 105071. [Google Scholar] [CrossRef]
- Qin, J.; Lu, J.; Cao, M.; Hu, C. Synthesis of porous CuO–CeO2 nanospheres with an enhanced low-temperature CO oxidation activity. Nanoscale 2010, 2, 2739–2743. [Google Scholar] [CrossRef] [PubMed]
- Kašpar, J.; Fornasiero, P.; Graziani, M. Use of CeO2-based oxides in the three-way catalysis. Catal. Today 1999, 50, 285–298. [Google Scholar] [CrossRef]
- Mai, H.-X.; Sun, L.-D.; Zhang, Y.-W.; Si, R.; Feng, W.; Zhang, H.-P.; Liu, H.-C.; Yan, C.-H. Shape-Selective Synthesis and Oxygen Storage Behavior of Ceria Nanopolyhedra, Nanorods, and Nanocubes. J. Phys. Chem. B 2005, 109, 24380–24385. [Google Scholar] [CrossRef]
- Lykaki, M.; Pachatouridou, E.; Iliopoulou, E.; Carabineiro, S.A.; Konsolakis, M. Impact of the synthesis parameters on the solid state properties and the CO oxidation performance of ceria nanoparticles. RSC Adv. 2017, 7, 6160–6169. [Google Scholar] [CrossRef] [Green Version]
- Sayle, T.X.T.; Parker, S.C.; Catlow, C.R.A. The role of oxygen vacancies on ceria surfaces in the oxidation of carbon monoxide. Surf. Sci. 1994, 316, 329–336. [Google Scholar] [CrossRef]
- Conesa, J. Computer modeling of surfaces and defects on cerium dioxide. Surf. Sci. 1995, 339, 337–352. [Google Scholar] [CrossRef]
- Wu, Z.; Li, M.; Overbury, S.H. On the structure dependence of CO oxidation over CeO2 nanocrystals with well-defined surface planes. J. Catal. 2012, 285, 61–73. [Google Scholar] [CrossRef]
- Konsolakis, M. The role of Copper-Ceria interactions in catalysis science: Recent theoretical and experimental advances. Appl. Catal. B Environ. 2016, 198, 49–66. [Google Scholar] [CrossRef]
- Konsolakis, M.; Lykaki, M. Recent Advances on the Rational Design of Non-Precious Metal Oxide Catalysts Exemplified by CuOx/CeO2 Binary System: Implications of Size, Shape and Electronic Effects on Intrinsic Reactivity and Metal-Support Interactions. Catalysts 2020, 10, 160. [Google Scholar] [CrossRef] [Green Version]
- Puigdollers, A.R.; Schlexer, P.; Tosoni, S.; Pacchioni, G. Increasing oxide reducibility: The role of metal/oxide interfaces in the formation of oxygen vacancies. ACS Catal. 2017, 7, 6493–6513. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Lai, Z.; Zhang, X.; Fan, Z.; He, Q.; Tan, C.; Zhang, H. Phase engineering of nanomaterials. Nat. Rev. Chem. 2020, 4, 243–256. [Google Scholar] [CrossRef]
- Konsolakis, M.; Lykaki, M. Facet-Dependent Reactivity of Ceria Nanoparticles Exemplified by CeO2-Based Transition Metal Catalysts: A Critical Review. Catalysts 2021, 11, 452. [Google Scholar] [CrossRef]
- Li, R.; Deng, H.; Zhang, X.; Wang, J.J.; Awasthi, M.K.; Wang, Q.; Xiao, R.; Zhou, B.; Du, J.; Zhang, Z. High-efficiency removal of Pb(II) and humate by a CeO2–MoS2 hybrid magnetic biochar. Bioresour. Technol. 2019, 273, 335–340. [Google Scholar] [CrossRef]
- Melchionna, M.; Bevilacqua, M.; Fornasiero, P. The electrifying effects of carbon-CeO2 interfaces in (electro)catalysis. Mater. Today Adv. 2020, 6, 1000502. [Google Scholar] [CrossRef]
- Khataee, A.; Gholami, P.; Kalderis, D.; Pachatouridou, E.; Konsolakis, M. Preparation of novel CeO2-biochar nanocomposite for sonocatalytic degradation of a textile dye. Ultrason. Sonochem. 2018, 41, 503–513. [Google Scholar] [CrossRef]
- Pi, L.; Jiang, R.; Cai, W.; Wang, L.; Wang, Y.; Cai, J.; Mao, X. Bionic Preparation of CeO2-Encapsulated Nitrogen Self-Doped Biochars for Highly Efficient Oxygen Reduction. ACS Appl. Mater. Interfaces 2020, 12, 3642–3653. [Google Scholar] [CrossRef]
- Inyang, M.; Dickenson, E. The potential role of biochar in the removal of organic and microbial contaminants from potable and reuse water: A review. Chemosphere 2015, 134, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Chen, J.; Yue, S.; Li, G. A comparative study of modified cotton biochar and activated carbon based catalysts in low temperature SCR. Fuel 2015, 156, 47. [Google Scholar] [CrossRef]
- Hasa, B.; Martino, E.; Vakros, J.; Trakakis, G.; Galiotis, C.; Katsaounis, A. Effect of carbon support on the electrocatalytic properties of Pt-Ru catalysts. Chem. Electron. Chem. 2019, 6, 4970–4979. [Google Scholar]
- Mian, M.; Liu, G. Recent progress in biochar-supported photocatalysts: Synthesis, role of biochar, and applications. RSC Adv. 2018, 8, 14237. [Google Scholar] [CrossRef] [Green Version]
- Vakros, J. Biochars and their use as transesterification catalysts for biodiesel production: A short review. Catalysts 2018, 8, 562. [Google Scholar] [CrossRef] [Green Version]
- Vakros, J.; Manariotis, I.D.; Dracopoulos, V.; Mantzavinos, D.; Lianos, P. Biochar from Spent Malt Rootlets and Its Application to an Energy Conversion and Storage Device. Chemosensors 2021, 9, 57. [Google Scholar] [CrossRef]
- Tan, X.F.; Liu, Y.G.; Gu, Y.L.; Xu, Y.; Zeng, G.M.; Hu, X.J.; Liu, S.B.; Wang, X.; Liu, S.M.; Li, J. Biochar-based nano-composites for the decontamination of wastewater: A review. Bioresour. Technol. 2016, 212, 318–333. [Google Scholar] [CrossRef] [PubMed]
- Grilla, E.; Vakros, J.; Konstantinou, I.; Manariotis, I.D.; Mantzavinos, D. Activation of Persulfate by Biochar from Spent Malt Rootlets for the Degradation of Trimethoprim in the Presence of Inorganic Ions. J. Chem. Technol. Biotechnol. 2020, 95, 2348–2358. [Google Scholar] [CrossRef]
- Lykoudi, A.; Frontistis, Z.; Vakros, J.; Manariotis, I.D.; Mantzavinos, D. Degradation of Sulfamethoxazole with Persulfate Using Spent Coffee Grounds Biochar as Activator. J. Environ. Manag. 2020, 271, 111022. [Google Scholar] [CrossRef]
- Magioglou, E.; Frontistis, Z.; Vakros, J.; Manariotis, I.; Mantzavinos, D. Activation of Persulfate by Biochars from Valorized Olive Stones for the Degradation of Sulfamethoxazole. Catalysts 2019, 9, 419. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Mozaz, S.; Vaz-Moreira, I.; Varela Della Giustina, S.; Llorca, M.; Barceló, D.; Schubert, S.; Berendonk, T.U.; Michael-Kordatou, I.; Fatta-Kassinos, D.; Martinez, J.L.; et al. Antibiotic residues in final effluents of European wastewater treatment plants and their impact on the aquatic environment. Environ. Int. 2020, 140, 105733. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G.J. Environmental factors influencing the development and spread of antibiotic resistance. FEMS Microbiol. Rev. 2018, 42, fux053. [Google Scholar] [CrossRef]
- Kemmou, L.; Frontistis, Z.; Vakros, J.; Manariotis, I.D.; Mantzavinos, D. Degradation of antibiotic sulfamethoxazole by biochar–activated persulfate: Factors affecting the activation and degradation processes. Catal. Today 2018, 313, 128–133. [Google Scholar] [CrossRef]
- Ntaflou, M.; Vakros, J. Transesterification activity of modified biochars from spent malt rootlets using triacetin. J. Clean. Prod. 2020, 259, 120931. [Google Scholar] [CrossRef]
- Bourikas, K.; Vakros, J.; Kordulis, C.; Lycourghiotis, A. Potentiometric Mass Titrations: Experimental and Theoretical Establishment of a New Technique for Determining the Point of Zero Charge (PZC) of Metal (Hydr)Oxides. J. Phys. Chem. B 2003, 107, 9441–9451. [Google Scholar] [CrossRef]
- Avramiotis, E.; Frontistis, Z.; Manariotis, I.D.; Vakros, J.; Mantzavinos, D. On the Performance of a Sustainable Rice Husk Biochar for the Activation of Persulfate and the Degradation of Antibiotics. Catalysts 2021, 11, 1303. [Google Scholar] [CrossRef]
- Tala, W.; Chantara, S. Use of spent coffee ground biochar as ambient PAHs sorbent and novel extraction method for GC-MS analysis. Environ. Sci. Pollut. Res. 2019, 26, 13025–13040. [Google Scholar] [CrossRef] [PubMed]
- Gulicovski, J.J.; Bračko, I.; Milonjić, S.K. Morphology and the isoelectric point of nanosized aqueous ceria sols. Mater. Chem. Phys. 2014, 148, 868–873. [Google Scholar] [CrossRef]
- Papavasiliou, J.; Rawski, M.; Vakros, J.; Avgouropoulos, G. A Novel Post-Synthesis Modification of CuO-CeO2 Catalysts: Effect on Their Activity for Selective CO Oxidation. ChemCatChem 2018, 10, 2096–2106. [Google Scholar] [CrossRef]
- Araujo, V.D.; Bellido, J.D.A.; Bernardi, M.I.B.; Assaf, J.M.; Assaf, E.M. CuO–CeO2 catalysts synthesized in one-step: Characterization and PROX performance. Int. J. Hydrogen Energy 2012, 37, 5498–5507. [Google Scholar] [CrossRef]
- Aguilab, G.; Guerrerob, S.; Arayaa, P. Effect of the preparation method and calcination temperature on the oxidation activity of CO at low temperature on CuO–CeO2/SiO2 catalysts. Appl. Catal. A 2013, 462–463, 56–63. [Google Scholar] [CrossRef]
- Rao, G.R.; Mishra, B.G. A comparative UV–vis-diffuse reflectance study on the location and interaction of cerium ions in Al- and Zr-pillared montmorillonite clays. Mater. Chem. Phys. 2005, 89, 110–115. [Google Scholar] [CrossRef]
- Sun, S.; Mao, D.; Yu, J. Enhanced CO oxidation activity of CuO/CeO2 catalyst prepared by surfactant-assisted impregnation method. J. Rare Earths 2015, 32, 1268–1274. [Google Scholar] [CrossRef]
- Lopez, J.M.; Gilbank, A.L.; Garcia, T.; Solsona, B.; Agouram, S.; Torrente-Murciano, L. The prevalence of surface oxygen vacancies over the mobility of bulk oxygen in nanostructured ceria for the total toluene oxidation. Appl. Catal. B 2015, 174–175, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Spanier, J.E.; Robinson, R.D.; Zhang, F.; Chan, S.-W.; Herman, I.P. Size-dependent properties of CeO2−y nanoparticles as studied by Raman scattering. Phys. Rev. B 2001, 64, 245407. [Google Scholar] [CrossRef] [Green Version]
- Wacławek, S.; Lutze, H.V.; Grübel, K.; Padil, V.V.T.; Cerník, M.; Dionysiou, D.D. Chemistry of persulfates in water and wastewater treatment: A review. Chem. Eng. J. 2017, 330, 44–62. [Google Scholar] [CrossRef]
- Schott, H.; Astigarrabia, E. Isoelectric points of some sulfonamides: Determination by microelectrophoresis and by calculations involving acid–base strength. J. Pharm. Sci. 1988, 77, 918–920. [Google Scholar] [CrossRef]
- Avisar, D.; Primor, O.; Gozlan, I.; Mamane, H. Sorption of sulfonamides and tetracyclines to montmorillonite clay. Water Air Soil Pollut. 2010, 209, 439–450. [Google Scholar] [CrossRef]
- Heo, J.; Yoon, Y.; Lee, G.; Kim, Y.; Han, J.; Park, C.M. Enhanced adsorption of bisphenol A and sulfamethoxazole by a novel magnetic CuZnFe2O4–biochar composite. Bioresour. Technol. 2019, 281, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Bancirova, M. Sodium azide as a specific quencher of singlet oxygen during chemiluminescent detection by luminol and Cypridina luciferin analogues. Luminescence 2011, 26, 685–688. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | T Calc (°C) | SSA (m2 g−1) | pzc | D (XRD) (nm) | % CeO2 Content | Eg (eV) | %O2 Uptake in TGA |
|---|---|---|---|---|---|---|---|
| BC/Ce-300 | 300 | 119 | 6.8 | 29.2 | 16 | 3.09 | 0 |
| BC/Ce-300-5 h | 300 | 110 | 6.7 | 20.7 | 14 | 3.10 | 0 |
| BC/Ce-350 | 350 | 126 | 6.5 | 29.8 | 22 | 3.07 | 1.7 |
| BC/Ce-400 | 400 | 69 | 3.0 | 18.9 | 7 | 3.10 | 0.5 |
| BC/Ce-500 | 500 | 14 | 3.0 | 16.0 | 2 | 3.12 | 2.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papatheodorou, G.; Ntzoufra, P.; Hapeshi, E.; Vakros, J.; Mantzavinos, D. Hybrid Biochar/Ceria Nanomaterials: Synthesis, Characterization and Activity Assessment for the Persulfate-Induced Degradation of Antibiotic Sulfamethoxazole. Nanomaterials 2022, 12, 194. https://doi.org/10.3390/nano12020194
Papatheodorou G, Ntzoufra P, Hapeshi E, Vakros J, Mantzavinos D. Hybrid Biochar/Ceria Nanomaterials: Synthesis, Characterization and Activity Assessment for the Persulfate-Induced Degradation of Antibiotic Sulfamethoxazole. Nanomaterials. 2022; 12(2):194. https://doi.org/10.3390/nano12020194
Chicago/Turabian StylePapatheodorou, Golfo, Paraskevi Ntzoufra, Evroula Hapeshi, John Vakros, and Dionissios Mantzavinos. 2022. "Hybrid Biochar/Ceria Nanomaterials: Synthesis, Characterization and Activity Assessment for the Persulfate-Induced Degradation of Antibiotic Sulfamethoxazole" Nanomaterials 12, no. 2: 194. https://doi.org/10.3390/nano12020194
APA StylePapatheodorou, G., Ntzoufra, P., Hapeshi, E., Vakros, J., & Mantzavinos, D. (2022). Hybrid Biochar/Ceria Nanomaterials: Synthesis, Characterization and Activity Assessment for the Persulfate-Induced Degradation of Antibiotic Sulfamethoxazole. Nanomaterials, 12(2), 194. https://doi.org/10.3390/nano12020194