
Attraction in Action: Reduction of Water to Dihydrogen Using Surface-Functionalized TiO2 Nanoparticles




Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Synthetic Procedures
2.2.1. TiO2 NPs Functionalization
2.2.2. Activation of Commercial P25 TiO2 NPs
2.2.3. Preparation of 1@TiO2
2.2.4. Ru@TiO2
2.2.5. Ru@TiO2 Using [Ru(bpy)2Cl2]
2.2.6. Rh@TiO2
2.2.7. rR-TiO2
2.2.8. RR-TiO2
2.2.9. Preparation of 1@ZrO2
2.2.10. Ru@ZrO2
2.2.11. rR-ZrO2
2.3. Dihydrogen Generation
2.3.1. General Procedure
2.3.2. Kinetic Measurements
2.3.3. Recycle Measurements
3. Results and Discussion
3.1. Ligand Functionalization, Surface Complexation and Material Characterisation
3.1.1. Anchoring Ligand Functionalization
3.1.2. Ligand Functionalized NP Characterisation Methods
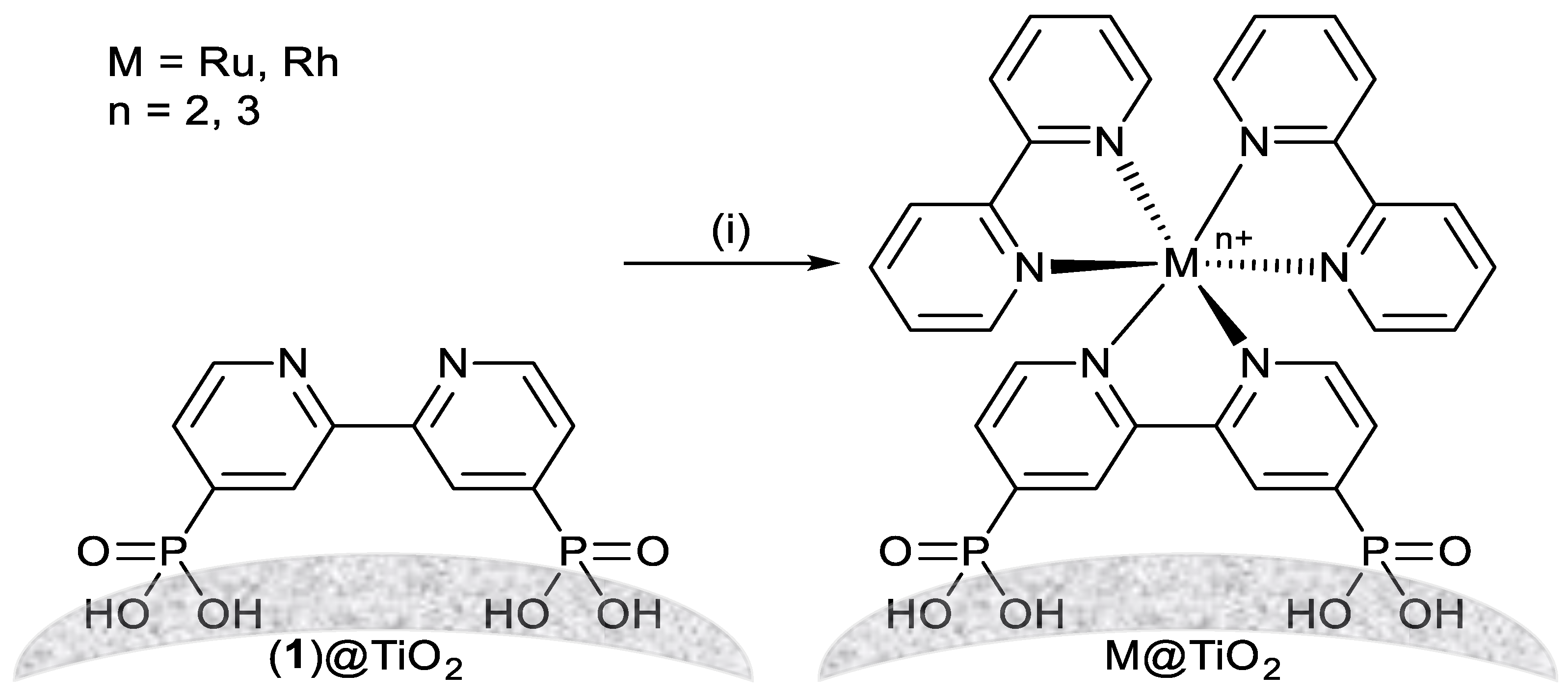
3.1.3. Nanoparticle Surface Complexation
3.1.4. Complex Functionalized Characterisation Methods
3.2. Dihydrogen Generation
3.2.1. Performance and Influence of Individual Components during Dihydrogen Generation
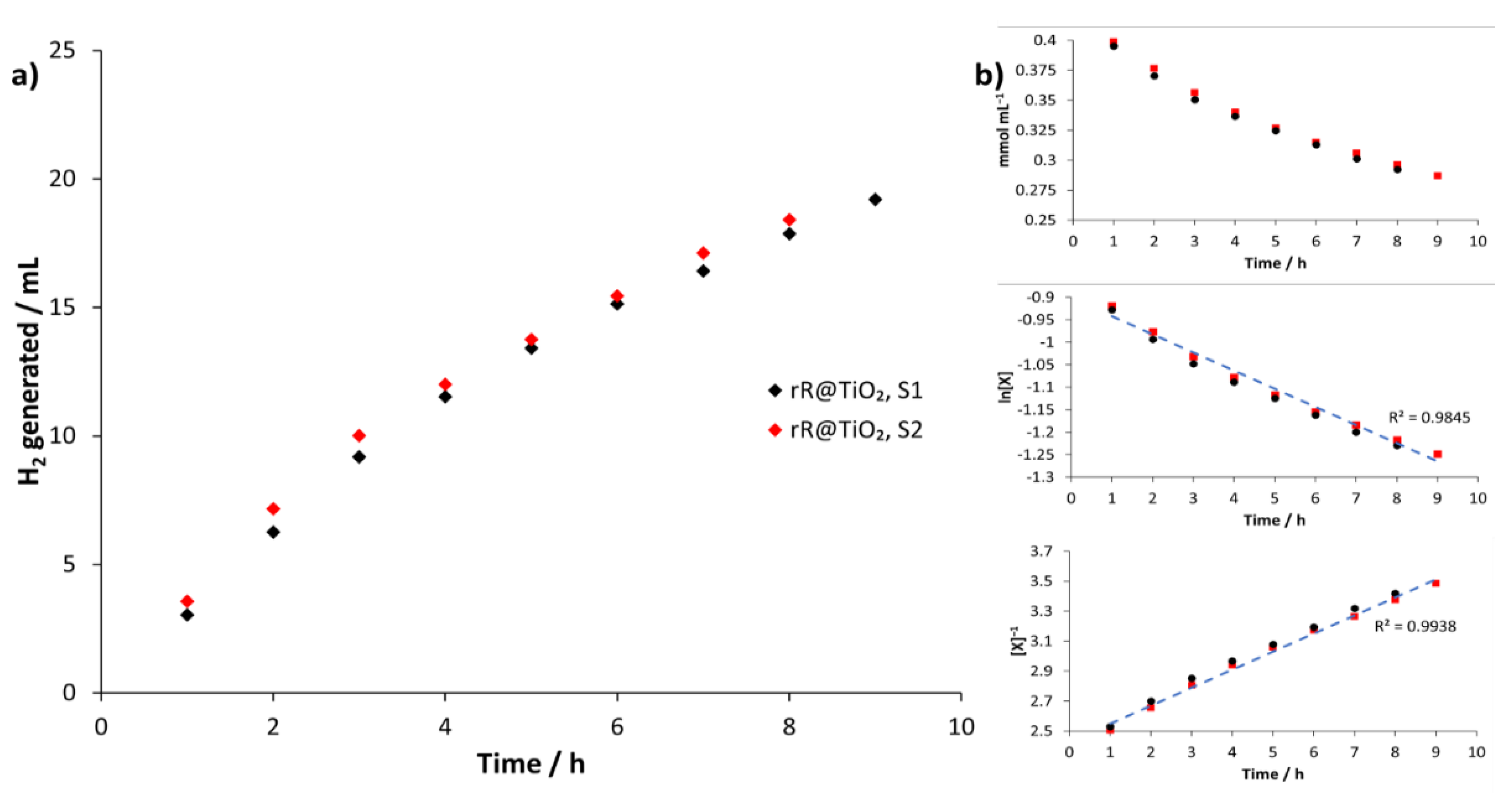
3.2.2. Kinetics of rR@TiO2 during Light Irradiation
3.2.3. Recyclability of rR@TiO2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Omer, A.M. Energy, environment and sustainable development. Renew. Sustain. Energy Rev. 2008, 12, 2265–2300. [Google Scholar] [CrossRef]
- Bölük, G.; Mert, M. Fossil & renewable energy consumption, GHGs (greenhouse gases) and economic growth: Evidence from a panel of EU (European Union) countries. Energy 2014, 74, 439–446. [Google Scholar] [CrossRef]
- Letcher, T.M. 1—Introduction With a Focus on Atmospheric Carbon Dioxide and Climate Change. In Future Energy: Improved, Sustainable and Clean Options for Our Planet, 3rd ed.; Letcher, T.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 3–17. [Google Scholar]
- Metzger, J.O.; Hüttermann, A. Sustainable global energy supply based on lignocellulosic biomass from afforestation of degraded areas. Sci. Nat. 2009, 96, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-N.; Wei, Y.-M.; Liu, L.-C.; Han, R.; Yu, B.-Y.; Wang, J.-W. Energy systems for climate change mitigation: A systematic review. Appl. Energy 2020, 263, 114602. [Google Scholar] [CrossRef]
- Rivard, E.; Trudeau, M.; Zaghib, K. Hydrogen Storage for Mobility: A Review. Materials 2019, 12, 1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.rsc.org/periodic-table/element/1/hydrogen (accessed on 14 June 2021).
- Timmerberg, S.; Kaltschmitt, M.; Finkbeiner, M. Hydrogen and Hydrogen-Derived Fuels through Methane Decomposition of Natural Gas–GHG Emissions and Costs; Elsevier: Amsterdam, The Netherlands, 2020; Volume 7, p. 100043. [Google Scholar]
- Wang, S.; Lu, A.; Zhong, C.-J. Hydrogen production from water electrolysis: Role of catalysts. Nano Converg. 2021, 8, 4. [Google Scholar] [CrossRef]
- Chen, L.; Qi, Z.; Zhang, S.; Su, J.; Somorjai, G.A. Catalytic Hydrogen Production from Methane: A Review on Recent Progress and Prospect. Catalysts 2020, 10, 858. [Google Scholar] [CrossRef]
- Dovì, V.G.; Friedler, F.; Huisingh, D.; Klemeš, J.J. Cleaner energy for sustainable future. J. Clean. Prod. 2009, 17, 889–895. [Google Scholar] [CrossRef]
- Johnson, T.C.; Morris, D.J.; Wills, M. Hydrogen generation from formic acid and alcohols using homogeneous catalysts. Chem. Soc. Rev. 2010, 39, 81–88. [Google Scholar] [CrossRef]
- Sajjadi, S.; Khataee, A.; Darvishi Cheshmeh Soltani, R.; Hasanzadeh, A. N, S co-doped graphene quantum dot–decorated Fe3O4 nanostructures: Preparation, characterization and catalytic activity. J. Phys. Chem. 2019, 127, 140–150. [Google Scholar] [CrossRef]
- Hassan, A.F.; Elhadidy, H. Effect of Zr+4 doping on characteristics and sonocatalytic activity of TiO2/carbon nanotubes composite catalyst for degradation of chlorpyrifos. J. Phys. Chem. 2019, 129, 180–187. [Google Scholar] [CrossRef]
- Maleki, A.; Taheri-Ledari, R.; Ghalavand, R.; Firouzi-Haji, R. Palladium-decorated o-phenylenediamine-functionalized Fe3O4/SiO2 magnetic nanoparticles: A promising solid-state catalytic system used for Suzuki–Miyaura coupling reactions. J. Phys. Chem. 2020, 136, 109200. [Google Scholar] [CrossRef]
- Stevens, P.D.; Fan, J.; Gardimalla, H.M.R.; Yen, M.; Gao, Y. Superparamagnetic Nanoparticle-Supported Catalysis of Suzuki Cross-Coupling Reactions. Org. Lett. 2005, 7, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Zecchina, A.; Bordiga, S.; Groppo, E. Selective Nanocatalysts and Nanoscience: Concepts for Heterogeneous and Homogeneous Catalysis; Wiley-VHC: Weinheim, Germany, 2011. [Google Scholar]
- Samira Bagheri, N.M.J. Nanocatalysts in Environmental Applications; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Albonetti, S.; Mazzoni, R.; Cavani, F. CHAPTER 1 Homogeneous, Heterogeneous and Nanocatalysis. In Transition Metal Catalysis in Aerobic Alcohol Oxidation; Green Chemistry Series; Cardona, F., Parmeggiani, C., Eds.; RSC: London, UK, 2015; pp. 1–39. [Google Scholar]
- Kumaravel, V.; Imam, M.D.; Badreldin, A.; Chava, R.K.; Do, J.Y.; Kang, M.; Abdel-Wahab, A. Photocatalytic Hydrogen Production: Role of Sacrificial Reagents on the Activity of Oxide, Carbon, and Sulfide Catalysts. Catalysts 2019, 9, 276. [Google Scholar] [CrossRef] [Green Version]
- Yoong, L.S.; Chong, F.K.; Dutta, B.K. Development of copper-doped TiO2 photocatalyst for hydrogen production under visible light. Energy 2009, 34, 1652–1661. [Google Scholar] [CrossRef]
- Kanade, K.G.; Kale, B.B.; Baeg, J.-O.; Lee, S.M.; Lee, C.W.; Moon, S.-J.; Chang, H. Self-assembled aligned Cu doped ZnO nanoparticles for photocatalytic hydrogen production under visible light irradiation. Mater. Chem. Phys. 2007, 102, 98–104. [Google Scholar] [CrossRef]
- Chowdhury, P.; Gomaa, H.; Ray, A.K. Sacrificial hydrogen generation from aqueous triethanolamine with Eosin Y-sensitized Pt/TiO2 photocatalyst in UV, visible and solar light irradiation. Chemosphere 2015, 121, 54–61. [Google Scholar] [CrossRef]
- Popugaeva, D.; Tian, T.; Ray, A.K. Hydrogen production from aqueous triethanolamine solution using Eosin Y-sensitized ZnO photocatalyst doped with platinum. Int. J. Hydrog. Energy 2020, 45, 11097–11107. [Google Scholar] [CrossRef]
- Zuo, F.; Wang, L.; Wu, T.; Zhang, Z.; Borchardt, D.; Feng, P. Self-Doped Ti3+ Enhanced Photocatalyst for Hydrogen Production under Visible Light. J. Am. Chem. Soc. 2010, 132, 11856–11857. [Google Scholar] [CrossRef]
- Romero, N.; Guerra, R.B.; Gil, L.; Drouet, S.; Salmeron-Sànchez, I.; Illa, O.; Philippot, K.; Natali, M.; García-Antón, J.; Sala, X. TiO2-mediated visible-light-driven hydrogen evolution by ligand-capped Ru nanoparticles. Sustain. Energy Fuels 2020, 4, 4170–4178. [Google Scholar] [CrossRef]
- Kirch, M.; Lehn, J.-M.; Sauvage, J.-P. Hydrogen Generation by Visible Light Irradiation of Aqueous Solutions of Metal Complexes. An approach to the photochemical conversion and storage of solar energy. Helv. Chim. Acta 1979, 62, 1345–1384. [Google Scholar] [CrossRef]
- Ansari, S.A.; Cho, M.H. Highly Visible Light Responsive, Narrow Band gap TiO2 Nanoparticles Modified by Elemental Red Phosphorus for Photocatalysis and Photoelectrochemical Applications. Sci. Rep. 2016, 6, 25405. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Sunaina; Yadav, K.K.; Bajpai, V.K.; Jha, M. Tuning the bandgap of m-ZrO2 by incorporation of copper nanoparticles into visible region for the treatment of organic pollutants. Mater. Res. Bull. 2020, 123, 110698. [Google Scholar] [CrossRef]
- Lenis-Rojas, O.A.; Fernandes, A.R.; Roma-Rodrigues, C.; Baptista, P.V.; Marques, F.; Pérez-Fernández, D.; Guerra-Varela, J.; Sánchez, L.; Vázquez-García, D.; Torres, M.L.; et al. Heteroleptic mononuclear compounds of ruthenium(ii): Synthesis, structural analyses, in vitro antitumor activity and in vivo toxicity on zebrafish embryos. Dalton Trans. 2016, 45, 19127–19140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Telo, J.P.; Liao, C.; Hightower, S.E.; Clennan, E.L. Experimental and Computational Studies of Nuclear Substituted 1,1‘-Dimethyl-2,2‘-Bipyridinium Tetrafluoroborates. J. Phys. Chem. A 2007, 111, 13567–13574. [Google Scholar] [CrossRef] [PubMed]
- Maerker, G.; Case, F.H. The Synthesis of Some 4,4’-Disubstituted 2,2’-Bipyridines1. J. Am. Chem. Soc. 1958, 80, 2745–2748. [Google Scholar] [CrossRef]
- Han, W.-S.; Han, J.-K.; Kim, H.-Y.; Choi, M.J.; Kang, Y.-S.; Pac, C.; Kang, S.O. Electronic Optimization of Heteroleptic Ru(II) Bipyridine Complexes by Remote Substituents: Synthesis, Characterization, and Application to Dye-Sensitized Solar Cells. Inorg. Chem. 2011, 50, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Montalti, M.; Wadhwa, S.; Kim, W.Y.; Kipp, R.A.; Schmehl, R.H. Luminescent Ruthenium(II) Bipyridyl−Phosphonic Acid Complexes: pH Dependent Photophysical Behavior and Quenching with Divalent Metal Ions. Inorg. Chem. 2000, 39, 76–84. [Google Scholar] [CrossRef]
- Norris, M.R.; Concepcion, J.J.; Glasson, C.R.K.; Fang, Z.; Lapides, A.M.; Ashford, D.L.; Templeton, J.L.; Meyer, T.J. Synthesis of Phosphonic Acid Derivatized Bipyridine Ligands and Their Ruthenium Complexes. Inorg. Chem. 2013, 52, 12492–12501. [Google Scholar] [CrossRef]
- Freimann, S.A.; Zare, D.; Housecroft, C.E.; Constable, E.C. The SALSAC approach: Comparing the reactivity of solvent-dispersed nanoparticles with nanoparticulate surfaces. Nanoscale Adv. 2020, 2, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Freimann, S.A.; Prescimone, A.; Housecroft, C.E.; Constable, E.C. Turning over on sticky balls: Preparation and catalytic studies of surface-functionalized TiO2 nanoparticles. RSC Adv. 2021, 11, 5537–5547. [Google Scholar] [CrossRef]
- The surface area-to-volume ratio of NPs with an average radius of 10.5 nm is 28%. Surface concentrations were calculated as 0.28 times the number of TiO2 formula equivalents. When describing experiments using functionalized NPs, the number of equivalents or moles refer to the estimated amount of anchoring ligand or complex bound to the surface using the above formula.
- Available online: https://www.aerosil.com/sites/lists/RE/DocumentsSI/TI-1243-Titanium-Dioxide-as-Photocatalyst-EN.pdf. (accessed on 5 April 2020).
- Cooke, M.W.; Santoni, M.-P.; Loiseau, F.; Hasenknopf, B.; Hanan, G.S. Energy transfer in rhodium–ruthenium dimer-of-dimer assemblies. Inorg. Chim. Acta 2017, 454, 208–215. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | CatA/ µmol | CatB/ µmol | Irr./ h | Yield H2/ µmol | TONA | TOFA/ h–1 | TONB | TOFB/ h–1 |
|---|---|---|---|---|---|---|---|---|
| CdS a [20] | 1730 | - | 6 | 283 | 0.2 | 0.0 | - | - |
| TiO2 p25 a [20] | 3130 | - | 6 | 62 | 0.0 | 0.0 | - | - |
| 0.25%Pt@TiO2 b [23] | 1.3 | - | 3 | 432 | 337 | 112.4 | - | - |
| 0.75%Pt@ZnO c [24] | 3.8 | - | 3 | 745 | 194 | 64.6 | - | - |
| Ru@RuO2PPTiO2-RuP d [26] | 0.6 | - | 10 | 111 | 176 | 17.6 | - | - |
| Ru(bpy)32+, Rh(bpy)33+ e [27] | 1.8 | 11.7 | 32 | 1359 | 748 | 23.4 | 116 | 3.6 |
| Entry | 1@TiO2 a | RuCl3 | RhCl3 | bpy |
|---|---|---|---|---|
| Ru@TiO2 | 1.0 eq. | 0.79 eq. | 0 | 2.0 eq. |
| Rh@TiO2 | 1.0 eq. | 0 | 0.79 eq. | 2.0 eq. |
| rR@TiO2 | 1.0 eq. | 0.04 eq. | 0.76 eq. | 2.0 eq. |
| RR@TiO2 | 1.0 eq. | 0.25 eq. | 0.54 eq. | 2.0 eq. |
| Sample | 47Ti Conc. µg/L | 47Ti Conc. RSD a | 101Ru Conc. µg/L | 101Ru Conc. RSD a | 103Rh Conc. µg/L | 103Rh Conc. RSD a | 89Y (ISTD) Conc. µg/L | 89Y (ISTD) Conc. RSD a |
|---|---|---|---|---|---|---|---|---|
| c-NPs | 16,756.5 | 6.1 | 0.1 | 17.7 | 0.1 | 5.5 | 145,600.3 | 4.0 |
| a-NPs | 21,460.7 | 5.6 | 0.3 | 127.0 | 0.3 | 106.3 | 150,634.5 | 4.6 |
| 1@TiO2 | 20,492.8 | 4.7 | 0.1 | 12.8 | 0.1 | 2.9 | 146,834.6 | 5.6 |
| Ru@TiO2 | 21,142.6 | 21.0 | 232.7 | 2.4 | 0.1 | 4.0 | 141,660.1 | 5.1 |
| Rh@TiO2 | 21,879.5 | 5.7 | 0.2 | 9.6 | 117.7 | 3.6 | 148,959.7 | 5.3 |
| rR@TiO2 | 20,350.4 | 2.6 | 22.2 | 3.1 | 89.6 | 2.5 | 142,814.2 | 5.4 |
| RR@TiO2 | 18,659.6 | 5.9 | 62.0 | 4.2 | 53.5 | 2.9 | 146,352.9 | 5.9 |
| rR@TiO2 b | 13,466.9 | 2.9 | 3.5 | 3.2 | 3.1 | 4.5 | 142,412.9 | 6.5 |
| Entry No. | NPs /µmol | Byp /µmol | pH | Time /h | GCI a /a. u. | H2 /mL (mL h−1) |
|---|---|---|---|---|---|---|
| 1 | Ru@TiO2/1.5 | 18.6 | 10 | 8 | 152,250 | 3.14 (0.39) |
| 2 | Rh@TiO2/9.3 | 18.6 | 10 | 8 | 199,140 | 4.11 (0.51) |
| 3 b | Ru@TiO2 + Rh@TiO2/9.7 | 18.6 | 10 | 8 | 255,530 | 5.27 (0.66) |
| 4 | rR@TiO2/9.7 | 18.6 | 10 | 8 | 451,170 | 9.30 (1.16) |
| 5 | rR@TiO2/9.7 | 18.6 | 7.5 | 4 | 455,940 | 9.40 (2.34) |
| 6 | rR@TiO2/9.7 | 0 | 7.5 | 4 | 332,280 | 6.85 (1.71) |
| 7 c | rR@TiO2/9.7 | 18.6 | 7.5 | 4 | 34,720 | 0.50 (0.13) |
| 8 | RR@TiO2/9.7 | 18.6 | 7.5 | 4 | 384,200 | 7.92 (1.98) |
| 9 d | RR@TiO2/13.0 | 18.6 | 7.5 | 4 | 398,860 | 8.23 (2.06) |
| 10 d | RR@TiO2/13.0 | 210 | 7.5 | 4 | 397,710 | 8.20 (2.05) |
| 11 e | a-NP | 18.6 | 7.5 | 4 | 138,600 | 2.86 (0.71) |
| 12 | 1@TiO2/12.2 | 18.6 | 7.5 | 4 | 134,050 | 2.76 (0.69) |
| 13 | 0 | 18.6 | 7.5 | 4 | 0 | 0.00 (0.00) |
| 14 f | rR@TiO2/9.7 | 18.6 | 7.5 | 4 | 533,930 | 11.0 (2.75) |
| 15 g | rR@TiO2/9.7 | 18.6 | 7.5 | 4 | 318,210 | 6.5 (1.64) |
| 16 f | rR@ZrO2/9.7 | 18.6 | 7.5 | 4 | 29,720 | 0.6 (0.15) |
| 17 h | rR@TiO2/9.7 | 0 | 7.5 | 4 | 88,360 | 1.82 (0.46) |
| Runtime/h | GC Integral a S1 /a. u. | H2 Generated S1 /mL | GC Integral a S2 /a. u. | H2 Generated S2 /mL |
|---|---|---|---|---|
| 1 | 146,050 | 3.04 | 171,190 | 3.57 |
| 2 | 154,530 | 3.22 | 172,650 | 3.60 |
| 3 | 143,060 | 2.92 | 136,850 | 2.85 |
| 4 | 112,380 | 2.34 | 95,670 | 1.99 |
| 5 | 90,960 | 1.89 | 83,770 | 1.75 |
| 6 | 82,450 | 1.72 | 81,480 | 1.70 |
| 7 | 61,900 | 1.29 | 80,410 | 1.67 |
| 8 | 69,260 | 1.44 | 62,050 | 1.29 |
| 9 | 52,740 | 1.33 | - | - |
| Cycle/N | GC Integral a/a. u. | NPs Runtime/h | H2/mL (mL h−1) |
|---|---|---|---|
| 0 | 455,940 | 4 | 9.40 (2.35) |
| 1 | 463,320 | 8 | 9.55 (2.38) |
| 2 | 372,850 | 12 | 7.68 (1.92) |
| 3 | 344,720 | 16 | 7.10 (1.77) |
| 4 | 277,740 | 20 | 5.73 (1.43) |
| 5 | 258,630 | 24 | 5.33 (1.33) |
| 6 | 257,160 | 28 | 5.30 (1.33) |
| 7 | 228,530 | 32 | 4.71 (1.18) |
| 8 | 209,710 | 36 | 4.32 (1.08) |
| 9 | 191,940 | 40 | 3.96 (0.99) |
| 10 | 181,590 | 44 | 4.75 (0.94) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freimann, S.A.; Housecroft, C.E.; Constable, E.C. Attraction in Action: Reduction of Water to Dihydrogen Using Surface-Functionalized TiO2 Nanoparticles. Nanomaterials 2022, 12, 789. https://doi.org/10.3390/nano12050789
Freimann SA, Housecroft CE, Constable EC. Attraction in Action: Reduction of Water to Dihydrogen Using Surface-Functionalized TiO2 Nanoparticles. Nanomaterials. 2022; 12(5):789. https://doi.org/10.3390/nano12050789
Chicago/Turabian StyleFreimann, Sven A., Catherine E. Housecroft, and Edwin C. Constable. 2022. "Attraction in Action: Reduction of Water to Dihydrogen Using Surface-Functionalized TiO2 Nanoparticles" Nanomaterials 12, no. 5: 789. https://doi.org/10.3390/nano12050789
APA StyleFreimann, S. A., Housecroft, C. E., & Constable, E. C. (2022). Attraction in Action: Reduction of Water to Dihydrogen Using Surface-Functionalized TiO2 Nanoparticles. Nanomaterials, 12(5), 789. https://doi.org/10.3390/nano12050789