
Rational Design of Non-Noble Metal Single-Atom Catalysts in Lithium–Sulfur Batteries through First Principles Calculations


Abstract
:
{kind=link}
{kind=link}
{kind=link}
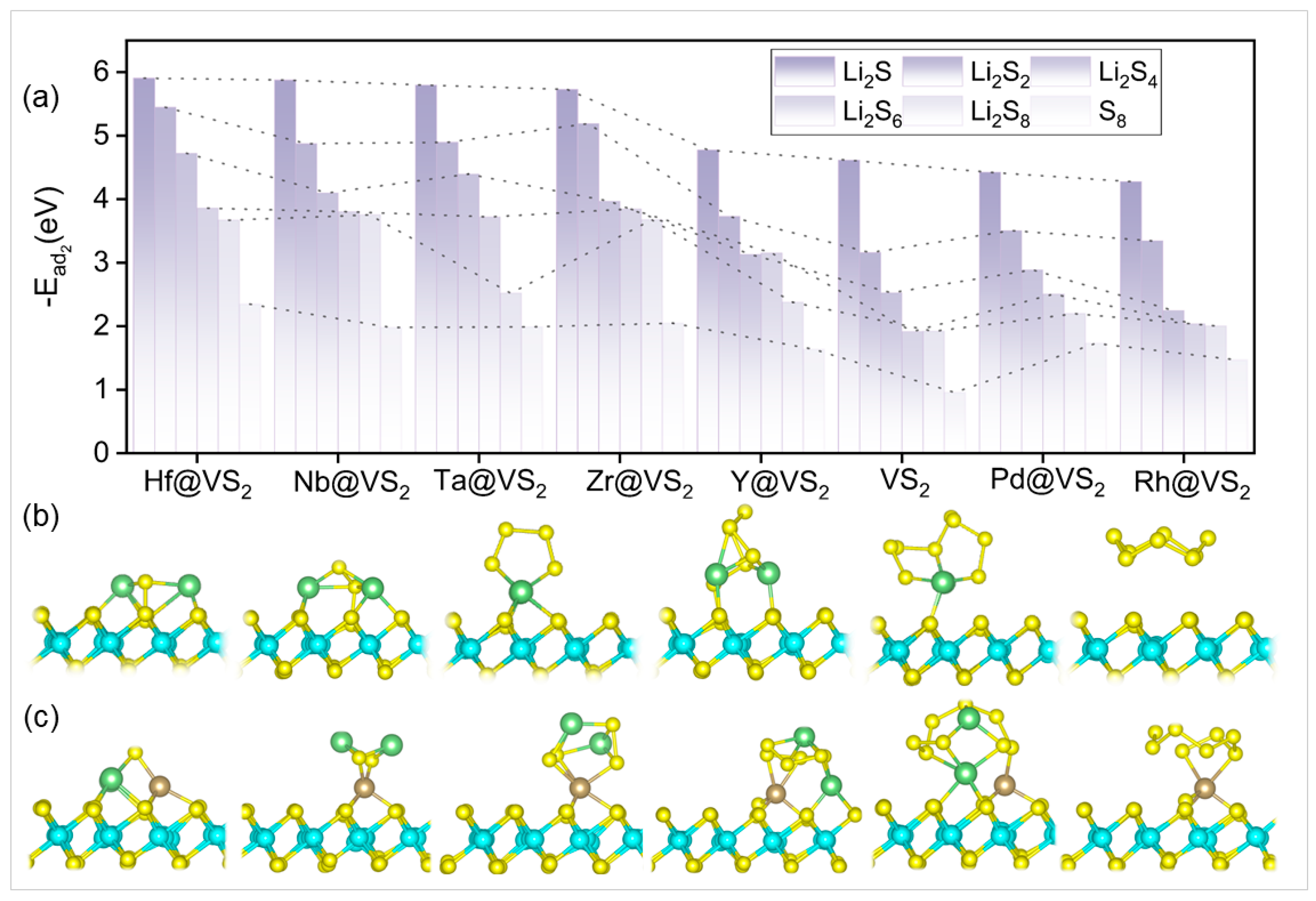{kind=link}
{kind=link}
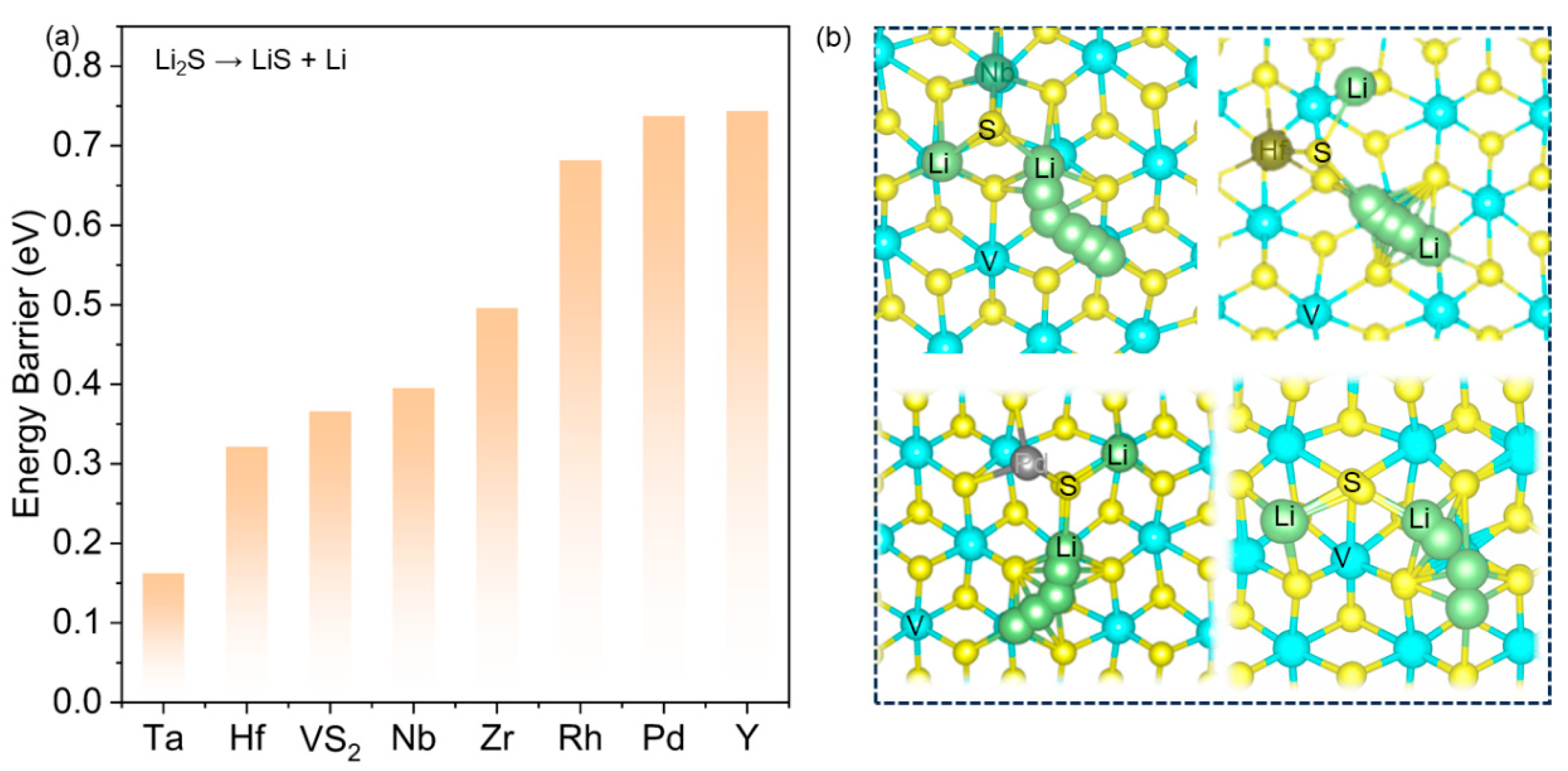{kind=link}
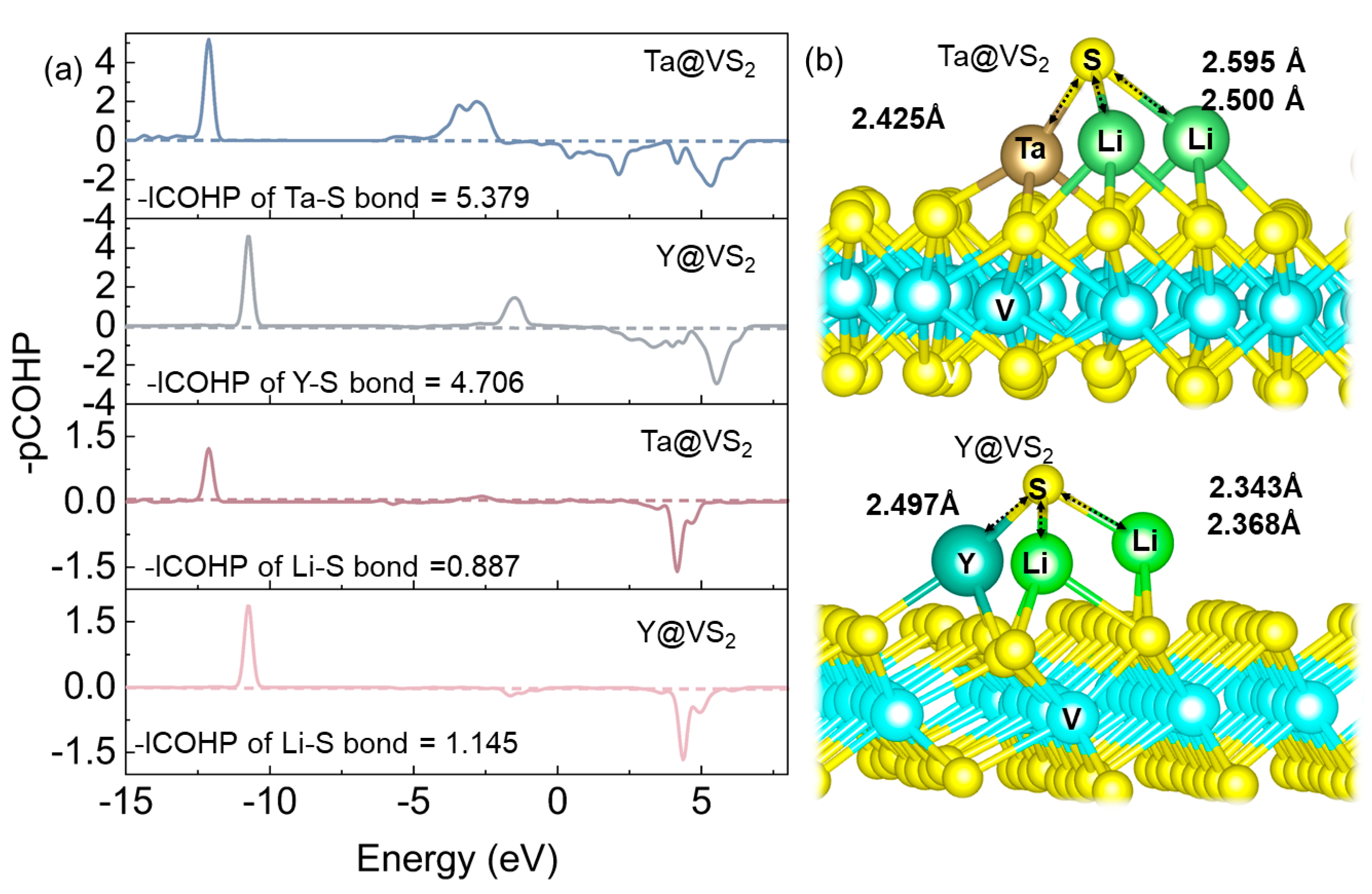{kind=link}
1. Introduction
2. Methods
3. Results and Discussion
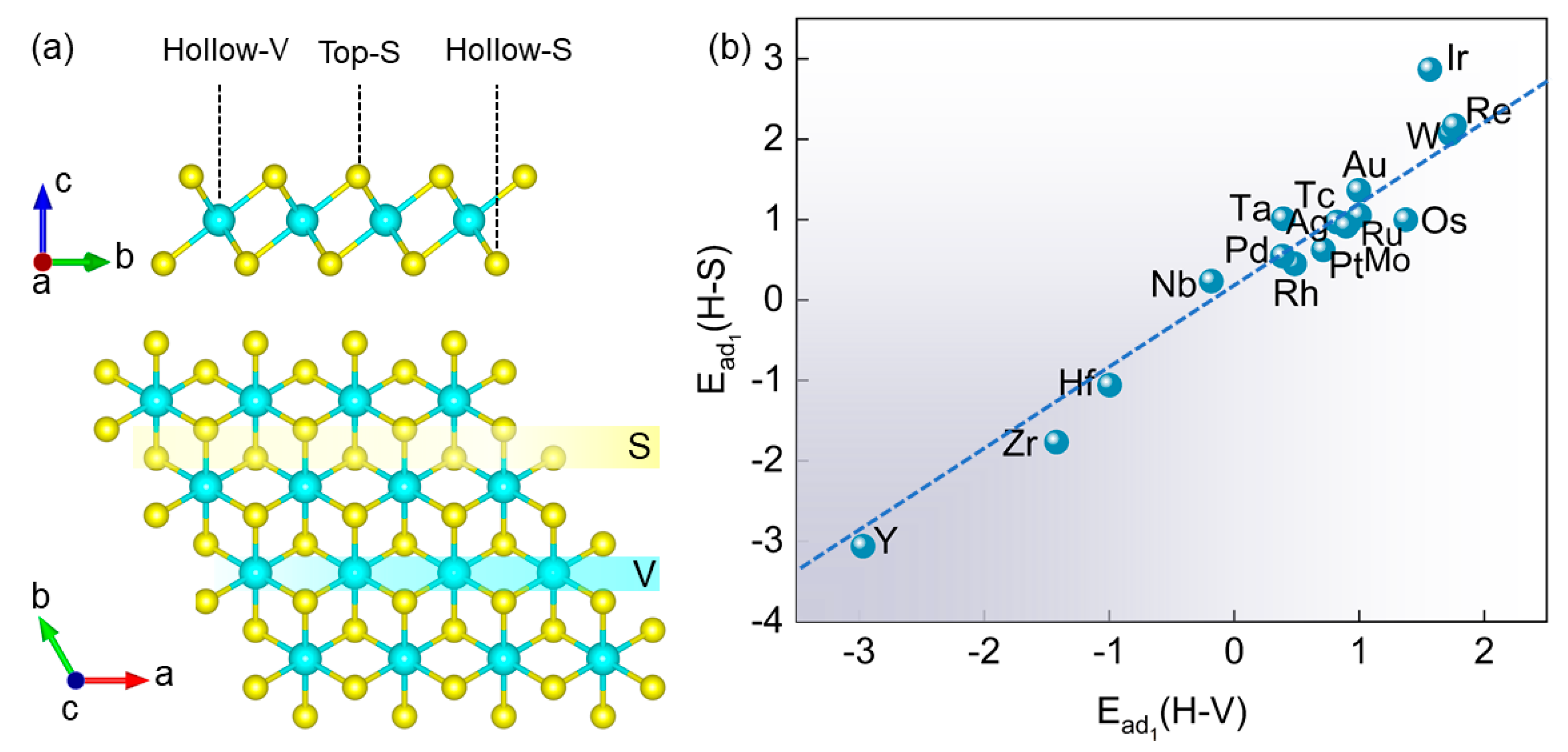
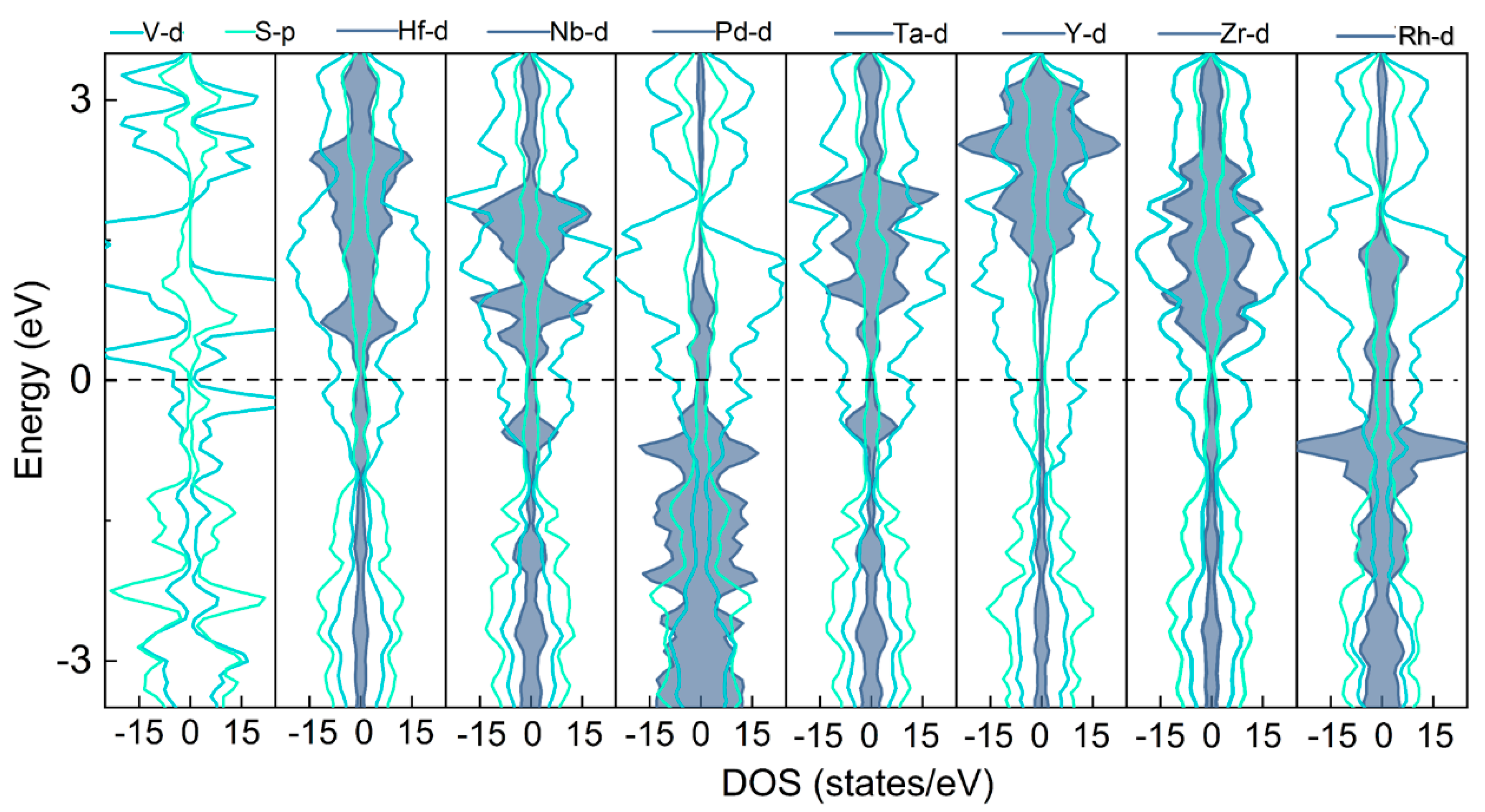
3.1. Structure and Stability of the TM@VS2 SACs
3.2. Anchoring Effect of TM@VS2 SACs
3.3. The Catalytic Mechanism of TM@VS2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bruce, P.G.; Freunberger, S.A.; Hardwick, L.J.; Tarascon, J.M. Li–O2 and Li–S batteries with high energy storage. Nat. Mater. 2012, 11, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hou, T.; Persson, K.A.; Zhang, Q. Combining theory and experiment in lithium–sulfur batteries: Current progress and future perspectives. Mater. Today 2019, 22, 142–158. [Google Scholar] [CrossRef]
- Evers, S.; Nazar, L.F. New Approaches for High Energy Density Lithium–Sulfur Battery Cathodes. Acc. Chem. Res. 2013, 46, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Demir-Cakan, R.; Morcrette, M.; Nouar, F.; Davoisne, C.; Devic, T.; Gonbeau, D.; Dominko, R.; Serre, C.; Férey, G.; Tarascon, J.-M. Cathode Composites for Li–S Batteries via the Use of Oxygenated Porous Architectures. J. Am. Chem. Soc. 2011, 133, 16154–16160. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Fu, Z.H.; Chen, X.; Zhang, Q. A review on theoretical models for lithium–sulfur battery cathodes. InfoMat 2022, 4, e12304. [Google Scholar] [CrossRef]
- Qian, X.; Jin, L.; Zhao, D.; Yang, X.; Wang, S.; Shen, X.; Rao, D.; Yao, S.; Zhou, Y.; Xiao, X. Ketjen Black-MnO Composite Coated Separator for High Performance Rechargeable Lithium-Sulfur Battery. Electrochim. Acta 2016, 192, 346–356. [Google Scholar] [CrossRef]
- Lu, Y.Q.; Wu, Y.J.; Sheng, T.; Peng, X.X.; Gao, Z.G.; Zhang, S.J.; Deng, L.; Nie, R.; Światowska, J.; Li, J.-T.; et al. Novel Sulfur Host Composed of Cobalt and Porous Graphitic Carbon Derived from MOFs for the High-Performance Li–S Battery. ACS Appl. Mater. Interfaces 2018, 10, 13499–13508. [Google Scholar] [CrossRef]
- Ji, X.; Lee, K.T.; Nazar, L.F. A highly ordered nanostructured carbon–sulphur cathode for lithium–sulphur batteries. Nat. Mater. 2009, 8, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Cao, Y.; Xiao, J.; Schwenzer, B.; Engelhard, M.H.; Saraf, L.V.; Nie, Z.; Exarhos, G.J.; Liu, J. A Soft Approach to Encapsulate Sulfur: Polyaniline Nanotubes for Lithium-Sulfur Batteries with Long Cycle Life. Adv. Mater. 2012, 24, 1176–1181. [Google Scholar] [CrossRef]
- Wang, L.; Hua, W.; Wan, X.; Feng, Z.; Hu, Z.; Li, H.; Niu, J.; Wang, L.; Wang, A.; Liu, J.Y. Design rules of a sulfur redox electrocatalyst for lithium–sulfur batteries. Adv. Mater. 2022, 34, 2110279. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, X.; Zhu, Z.; Zhong, Y.; Bando, Y.; Golberg, D.; Yao, J.; Wang, X. The Role of Geometric Sites in 2D Materials for Energy Storage. Joule 2018, 2, 1075–1094. [Google Scholar] [CrossRef]
- Chang, C.; Chen, W.; Chen, Y.; Chen, Y.; Chen, Y.; Ding, F.; Fan, C.; Fan, H.J.; Fan, Z.; Gong, C. Recent progress on two-dimensional materials. Acta Phys. -Chim. Sin. 2021, 37, 2108017. [Google Scholar] [CrossRef]
- Andritsos, E.I.; Lekakou, C.; Cai, Q. Single-Atom Catalysts as Promising Cathode Materials for Lithium–Sulfur Batteries. J. Phys. Chem. C 2021, 125, 18108–18118. [Google Scholar] [CrossRef]
- Su, Y.S.; Manthiram, A. Lithium–sulphur batteries with a microporous carbon paper as a bifunctional interlayer. Nat. Commun. 2012, 3, 1166. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Rangom, Y.; Kwok, C.Y.; Pang, Q.; Nazar, L.F. Interwoven MXene nanosheet/carbon-nanotube composites as Li–S cathode hosts. Adv. Mater. 2017, 29, 1603040. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ma, S.; Cai, H.; Zhou, H.; Huang, Z.; Hou, Z.; Wu, J.; Yang, W.; Yi, H.; Fu, C. Ultra-thin Fe3C nanosheets promote the adsorption and conversion of polysulfides in lithium-sulfur batteries. Energy Storage Mater. 2019, 18, 338–348. [Google Scholar] [CrossRef]
- Sim, E.S.; Yi, G.S.; Je, M.; Lee, Y.; Chung, Y.C. Understanding the anchoring behavior of titanium carbide-based MXenes depending on the functional group in LiS batteries: A density functional theory study. J. Power Sources 2017, 342, 64–69. [Google Scholar] [CrossRef]
- Ghazi, Z.A.; He, X.; Khattak, A.M.; Khan, N.A.; Liang, B.; Iqbal, A.; Wang, J.; Sin, H.; Li, L.; Tang, Z. MoS2/celgard separator as efficient polysulfide barrier for long-life lithium–sulfur batteries. Adv. Mater. 2017, 29, 1606817. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Xiao, Z.; Pan, H.; Wang, S.; Wang, R. Elastic Sandwich-Type rGO–VS2/S Composites with High Tap Density: Structural and Chemical Cooperativity Enabling Lithium–Sulfur Batteries with High Energy Density. Adv. Energy Mater. 2018, 8, 1702337. [Google Scholar] [CrossRef]
- Yang, X.F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Han, Z.; Zhao, S.; Xiao, J.; Zhong, X.; Sheng, J.; Lv, W.; Zhang, Q.; Zhou, G.; Cheng, H.M. Engineering d-p orbital hybridization in single-atom metal-embedded three-dimensional electrodes for Li–S batteries. Adv. Mater. 2021, 33, 2105947. [Google Scholar] [CrossRef]
- Lin, H.; Yang, L.; Jiang, X.; Li, G.; Zhang, T.; Yao, Q.; Zheng, G.W.; Lee, J.Y. Electrocatalysis of polysulfide conversion by sulfur-deficient MoS 2 nanoflakes for lithium–sulfur batteries. Energy Environ. Sci. 2017, 10, 1476–1486. [Google Scholar] [CrossRef]
- Yuan, Z.; Peng, H.J.; Hou, T.Z.; Huang, J.Q.; Chen, C.M.; Wang, D.W.; Cheng, X.B.; Wei, F.; Zhang, Q. Powering lithium–sulfur battery performance by propelling polysulfide redox at sulfiphilic hosts. Nano Lett. 2016, 16, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Liang, J.; Ye, S.; Zhang, Q.; Liu, J. Fundamental, application and opportunities of single atom catalysts for Li-S batteries. Energy Storage Mater. 2023, 55, 322–355. [Google Scholar] [CrossRef]
- Boyjoo, Y.; Shi, H.; Olsson, E.; Cai, Q.; Wu, Z.S.; Liu, J.; Lu, G.Q. Molecular-level design of pyrrhotite electrocatalyst decorated hierarchical porous carbon spheres as nanoreactors for lithium–sulfur batteries. Adv. Energy Mater. 2020, 10, 2000651. [Google Scholar] [CrossRef]
- Du, Z.; Chen, X.; Hu, W.; Chuang, C.; Xie, S.; Hu, A.; Yan, W.; Kong, X.; Wu, X.; Ji, H.; et al. Cobalt in nitrogen-doped graphene as single-atom catalyst for high-sulfur content lithium–sulfur batteries. J. Am. Chem. Soc. 2019, 141, 3977–3985. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Seh, Z.W.; Fu, Z.; Zhang, R.; Cui, Y.l. Understanding the anchoring effect of two-dimensional layered materials for lithium–sulfur batteries. Nano Lett. 2015, 15, 3780–3786. [Google Scholar] [CrossRef]
- He, J.; Hartmann, G.; Lee, M.; Hwang, G.S.; Chen, Y.; Manthiram, A. Freestanding 1T MoS2/graphene heterostructures as a highly efficient electrocatalyst for lithium polysulfides in Li–S batteries. Energy Environ. Sci. 2019, 12, 344–350. [Google Scholar] [CrossRef]
- Ren, Y.; Wang, B.; Liu, H.; Wu, H.; Bian, H.; Ma, Y.; Lu, H.; Tang, S.; Meng, X. CoP nanocages intercalated MXene nanosheets as a bifunctional mediator for suppressing polysulfide shuttling and dendritic growth in lithium-sulfur batteries. Chem. Eng. J. 2022, 450, 138046. [Google Scholar] [CrossRef]
- Park, J.; Yu, B.C.; Park, J.S.; Choi, J.W.; Kim, C.; Sung, Y.E.; Goodenough, J.B. Tungsten disulfide catalysts supported on a carbon cloth interlayer for high performance Li–S battery. Adv. Energy Mater. 2017, 7, 1602567. [Google Scholar] [CrossRef]
- Lin, H.; Zhang, S.; Zhang, T.; Cao, S.; Ye, H.; Yao, Q.; Zheng, G.W.; Lee, J.Y. A cathode-integrated sulfur-deficient Co9S8 catalytic interlayer for the reutilization of “lost” polysulfides in lithium–sulfur batteries. ACS Nano 2019, 13, 7073–7082. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, W.; Fan, H.; Cheng, F.; Su, D.; Wang, G. Promoting lithium polysulfide/sulfide redox kinetics by the catalyzing of zinc sulfide for high performance lithium-sulfur battery. Nano Energy 2018, 51, 73–82. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J.J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. Catalysis by supported single metal atoms. ACS Catal. 2017, 7, 34–59. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, L.; Ge, Q.; Chen, R.; Ni, M.; Utetiwabo, W.; Zhang, X.; Yang, W. Atomic Iron Catalysis of Polysulfide Conversion in Lithium–Sulfur Batteries. ACS Appl. Mater. Interfaces 2018, 10, 19311–19317. [Google Scholar] [CrossRef]
- Zeng, P.; Yuan, C.; Liu, G.; Gao, J.; Li, Y.; Zhang, L. Recent progress in electronic modulation of electrocatalysts for high-efficient polysulfide conversion of Li-S batteries. Chin. J. Catal. 2022, 43, 2946–2965. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Zhou, L.; Danilov, D.L.; Qiao, F.; Wang, J.; Li, H.; Eichel, R.A.; Notten, P.H. Sulfur reduction reaction in lithium–sulfur batteries: Mechanisms, catalysts, and characterization. Adv. Energy Mater. 2022, 12, 2202094. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Liu, Y.; Zhang, J.; Wang, D.; Xu, J. Rational Design of Non-Noble Metal Single-Atom Catalysts in Lithium–Sulfur Batteries through First Principles Calculations. Nanomaterials 2024, 14, 692. https://doi.org/10.3390/nano14080692
Li Y, Liu Y, Zhang J, Wang D, Xu J. Rational Design of Non-Noble Metal Single-Atom Catalysts in Lithium–Sulfur Batteries through First Principles Calculations. Nanomaterials. 2024; 14(8):692. https://doi.org/10.3390/nano14080692
Chicago/Turabian StyleLi, Yang, Yao Liu, Jinhui Zhang, Dashuai Wang, and Jing Xu. 2024. "Rational Design of Non-Noble Metal Single-Atom Catalysts in Lithium–Sulfur Batteries through First Principles Calculations" Nanomaterials 14, no. 8: 692. https://doi.org/10.3390/nano14080692
APA StyleLi, Y., Liu, Y., Zhang, J., Wang, D., & Xu, J. (2024). Rational Design of Non-Noble Metal Single-Atom Catalysts in Lithium–Sulfur Batteries through First Principles Calculations. Nanomaterials, 14(8), 692. https://doi.org/10.3390/nano14080692