This section may be divided by subheadings. It should provide a concise and precise description of the experimental results, their interpretation as well as the experimental conclusions that can be drawn.
3.1. Understanding the Phase Composition in CaO-Al2O3 System as a Function of the Temperature and Atmosphere Conditions
As a first approximation, we evaluated the vast synthesis conditions to facilitate the formation of the desired crystalline structure. For this, the CAO phosphors were synthesized employing the molten salt method by being heated from 1000 to 1400 °C for 2 h in an air atmosphere, employing a salt/CAO molar ratio of 3:1 and an Al
2O
3/CaO ratio of 1, using alumina of an average particle size d
50~6 μm. Regarding the amount of salt to be used, it is usually comprised between 80 and 120% in the weight of the reactant mixture, or using molar ratio salt/complex oxide (S/O), S/O = 1:1, 3:1, 4:1, 5:1 or 20:1 [
15]. After an experiment series, we found that the ratio 3:1 offers the most suitable synthesis conditions, analogously to synthesis for the SrAl
2O
4 phosphor.
Figure 1 shows the XRD pattern of the powder synthesized at 1000, 1100, 1200 and 1400 °C in the air atmosphere. At 1000 °C, the XRD shows characteristic peaks of the Ca
12Al
14O
33 cubic polymorph, whose pattern is characterized by four peaks centered in the 2θ axis at 18.11°, 27.82°, 33.39° and 41.20°, ascribed to the (211), (321), (420), (521) facet diffraction of the cubic, and matched with the Ca
12Al
14O
33 standard values given in JCPDS (No. 09-0413 or 70-2144) and their coexistence with the NaCl (JCPDF file 72-1668) and KCl (JCPDF file 76-3376) phases. From the CaO-Al
2O
3 diagram [
16], the Ca
12Al
14O
33 phase has the lowest forming temperature, around 1400 °C. Applying the molten salt synthesis, this forming temperature can be reduced down to be around 1000 °C, as is shown in
Figure 1. In addition, the XRD reveals the presence of another phase, which could be identified as Al
2O
3 (JCPDF file 73-1512). The intensity of the XRD peaks of the salt decreases with an increase in temperature, due to its vaporization during the thermal treatment. As commented above, the melting point of the eutectic mixture is observed at 659 °C; however, the remaining salts could be related to the presence of pure salts into the melt. In theory, the 100% weight loss of the eutectic mixture occurs at ca. 1010 °C, but a shift to a higher temperature is expected when other compounds are added into the system, as it has already reported in a previous work [
12].
At 1100 °C, the main phase Ca
12Al
14O
33 coexists with the monoclinic CaAl
2O
4 phase (JCPDS 70-0134) characterized by the diffraction peaks at 2θ = 18.99°, 30.14°, 35.42°, and 37.179°, attributed to the (−112), (220), (006), (313) crystal facets. At 1200 °C, the amount of monoclinic CaAl
2O
4 phase increases, reaching the maximum of CaAl
2O
4 fraction at 1400 °C. Taking into account the reaction mechanism established previously [
12], the most probable origin of the rest of these raw materials can be related to an unreacted Al
2O
3 core. From these observations, it can be stated that a thermal treatment at 1000 °C produces the Ca
12Al
14O
33 compound, and the CaAl
2O
4 compound is synthesized at 1400 °C. Between 1000 and 1400 °C, there is a coexistence of both phases. In the case of calcium aluminate host materials, it is difficult to produce the pure phase products owing to the generation of many phases together during the preparation, so the presence of the mixed phases may influence the luminescence performance of rare earth compounds.
In order to obtain a phosphorescence response, europium and neodymium were incorporated as the dopants to synthesize CAO: Eu
2+, Nd
3+ materials. The powders were thermally treated at 1000, 1200 and 1400 °C for 2 h in a furnace under a nitrogen–hydrogen (90N
2–10H
2) atmosphere to reduce Eu
3+ to Eu
2+. As shown in
Figure 2, the XRD pattern of the powder, which was thermally treated at 1000 °C for 2 h, is similar to that annealed in air. In both cases the main phase is Ca
12Al
14O
33. By contrast, under a reducing atmosphere, there is no residual salt revealed. Taking into account in the thermogravimetric (TG) analyses presented in detail elsewhere [
12], the salt starts to vaporize above the m.p. (659 °C), and it is expected that the 100% weight loss of the eutectic mixture occurs at ca. 1010 °C. However, the other components in the system can delay the total evaporation of the salt. During the thermal treatment of samples, a continuous gas flow of 90N
2-10H
2 at the pressure of 1 bar is used in a tubular furnace.
Thus, it may be expected that the gas flow passing through the chamber promotes the dragging of the salt during the processing time, removing the remains of salt when CaO and Al2O3 are incorporated. At 1200 °C, there is a coexistence of Ca12Al14O33 and CaAl2O4 phases with the minor amount of alumina Al2O3. Increasing the temperature up to 1400 °C results in the formation of the CaAl2O4 phase as a principal phase accompanying a trace amount of the Al2O3 phase. The crystallite size was also calculated; being 44(2), 55(3) and 63(3) nm for the samples synthesized at 1000, 1200 and 1400 °C, respectively. As expected, the crystallite size increases as the sintering temperature increases, indicating that the crystallinity of the CAO phase is increased.
Therefore, the development of Ca12Al14O33 and CaAl2O4 phases by the molten salt route at 1000 and 1400 °C, respectively, can be stated. Both polymorphs are the potential candidates to emit in the blue range conditioned by the successful incorporation of the doping elements.
3.2. Effect of the Heat Treatment on the Morphology, and Particle Size Distribution
Figure 3a–c depict the FE-SEM micrographs of the synthesized CAO particles, obtained from 1000 to 1400 °C. The morphology of the platelet-like shaped alumina micro-particles is preserved at all temperatures of treatment. The alumina acts as a template, promoting the dissolution−diffusion transport mechanism of calcia and the rare-earth dopants by the eutectic nature of the salt mixture [
12]. The procedure described allows synthesizing particles to possess a particle size in the micrometric range. Specifically, the particle size refers to the platelet-like shaped CAO microparticles. Due to the powder being not monodisperse, the particle size distribution describes the poly-dispersity character, reflecting the agglomeration state of these microparticles (see
Figure 3d–f, and
Table 1). The microparticles are also nanostructured, and the synthesis of CAO: Eu, Nd occurs at the surface. The eutectic flux dissolves the CaO and transports this reactant to the surface of Al
2O
3. These platelets-like particles have a particle size smaller than 10 µm for the samples synthesized at 1000 and 1200 °C and ca. 17 µm for the sample obtained at 1400 °C that exhibits an additional formation of sintering necks between platelet particles through the CAO phase.
These nanostructured platelet-like particles are composed of grains or primary particles, which evolve as a function of the synthesis temperature as a consequence of the coalescence and grain growth process, as is shown in
Figure 3. At 1000 °C the average grain size is ranged 100–500 nm, and a second phase is located between the grains.
At 1200 °C the average grain size increases up to 800–1200 nm, and in this case, the shape of the grains approaches straight grain boundaries and triple point junctions with 120° indicating a near-equilibrium microstructure. However, the heat treatment at 1400 °C promotes the grain growth in each individual microparticle to get grains ranging from 1 up to 2 µm. The preferential growth occurs in the platelet plane because the thickness of the CAO platelets does not exceed 2 µm, which is a beneficial dimension for applications. As the main phase at a higher temperature is CaAl
2O
4, it is possible to correlate the grain growth in each individual microparticle with phase evolution. The crystallite size described in the previous section also increases as the sintering temperature increases, which is in good agreement with the tendency observed by FE-SEM (
Figure 3). This indicates that the synthesized calcium aluminate phase (CAO) at the alumina microparticle surface is composed of crystalline material, forming polycrystalline particles since the calculated crystallite sizes are considerably smaller than the observed average grain sizes.
However, the presence of rare earth could play a relevant role in the nanostructure evolution. In this sense, the luminescence response will provide further pieces of evidence of the doping evolution and the micro-nano structure of each microparticle.
3.3. Photoluminescence Characterization and After Glow Properties
Figure 4a shows the PL emission spectrum of the particles synthesized in the N
2-H
2 atmosphere. Eu dopant was incorporated as Eu
2O
3, in its trivalent oxidation state. Different techniques are described in the literature to reduce Eu
3+ to Eu
2+, and their stabilization is a non-trivial task. One possibility is to exploit the most commonly used H
2 as a reduction agent. In our case, the annealing process carried out under a reductive atmosphere of 90N
2-10H
2 promotes the reduction of Eu
3+ to Eu
2+. Based on PL emission spectroscopy, it is straightforward to assess the presence of Eu
2+ and Eu
3+ emission centers. The Eu
3+ emission is characterized by the narrow lines between 550 and 750 nm attributed to 4f→4f (
5D
0/
7F
j = 0,1,2,3,4) transitions. As shown in
Figure 4a, the particles synthesized exhibit a broad emission band centered at 440 nm attributed to typical 4f
65d
1→4f
7 transitions of Eu
2+ under the excitation at 365 nm. A shift in the luminescence band position related to the Ca
12Al
14O
33 phase synthesized at 1000 °C can be explained by a small change in the crystal field effect on the Eu
2+ ions because of the splitting. It is important to remark that the emission peak attributed to the Eu
2+ cations [
17] transition becomes more intense with an increase in the temperature (from 1000 to 1400 °C). The emission intensities are lower for the powder synthesized at 1000 °C, where the cubic phase Ca
12Al
14O
33 is the predominant phase. At 1200 and 1400 °C, the emission intensity further increases. At 1400 °C, the single monoclinic phase of CaAl
2O
4 is steadily developed. As a first approach, the increment of the emission luminescence can be related to the phase composition. The inset in
Figure 4a shows the excitation spectrum monitored at the 440 nm wavelength. This spectrum covers a broad spectral region from 273 to 418 nm, assuming that the phosphor can be activated in this range. The standard solar spectrum (ASTM E-490) is presented to check that effectively a range of energies (UV-B and UV-A) of the solar irradiation spectrum can be used to stimulate the particles. It is known that the maximum fraction effectively used by Si solar cells [
18] is lower than the maximum fraction available in this range. Therefore, the employment of CAO: Eu, Nd material on the top of the solar panel can serve as persistent storage. [
19,
20] A comparison between the intensity obtained in CAO:Eu, Nd material and the commercial powder based on SrAl
2O
4:Eu, Dy (from Jinan Chenghao Technology Co., Ltd., Mingshihaoting, NO.12406 Jingshi East Road, Jinan, 250014, China) as received is shown in the Supporting Information (
Figure S1).
Besides the intense and broad photoluminescent emission, the CAO: Eu, Nd particles demonstrate a long-lasting luminescence after the stoppage of UV irradiation. The density of trapped carriers plays a key role in the enhancement of the afterglow duration. Specifically, co-doping with Nd
3+ results in a higher density of trapped carriers as compared with other rare earths [
4,
21].
Therefore, to prolong the afterglow, there must be empty energy levels available at shallow as well as deep locations in the bandgap. By the molten salt synthesis proposed here, the host can hold the luminescent center introduced, creating the required and distributed empty defects to prolong the blue luminescence.
Measurements of the decay curve are one of the most useful ways to determine the duration of the luminescence.
Figure 4b shows the persistent luminescence with decay time lasting from 0 s to 3600 s (1 h). The CAO: Eu, Nd particles were excited for 10 min, after that the excitation was cut off, and the afterglow curve was acquired. The profiles of the decay curves of the samples exhibit similar behavior. A multi-exponential decay profile was used to fit the experimental long afterglow decay data. Specifically, the observed afterglow decay curve was fitted by a quatri-exponential decay, following this equation:
where
I1 is the final intensity;
A1,
A2, A3, and
A4 are constants;
t is the decay time; and
τ1,
τ2,
τ3, and
τ4 are fitting parameters related to the decay rate of the phosphors. The fitting parameters are given in
Table 2.
The initial intensity of the sample thermally treated at 1000 °C is lower, and the intensity drops two orders of magnitude compared with the samples annealed at 1200 °C and 1400 °C for 1 h. This behavior can be related to the phase composition obtained in each thermal treatment. The presence of the monoclinic polymorph phase of CaAl
2O
4 at 1200 °C and 1400 °C has more ability to store and release the energy in comparison with Ca
12Al
14O
33. These four temporal processes may be related to different types of trap centers or different levels of the same trap species. As the persistent mechanism of CAO: Eu, Nd is not yet established, to evaluate the decay rate of the fast (
τ1), rapid (
τ2), medium (
τ3) and slow (
τ4) exponential decay components, the steady-state contributions are quantified with
Ai ×
τi products (
i = 1 to 4).
Ai represents the amplitude of the exponential decay and
τi, the lifetime [
22]. These values are acquired from the numerical fits, and by these calculations, the fractional contribution of each component can be obtained, according to the following expression:
The fraction of each component in the multi-exponential decay is:
The contributions of medium (τ3) and slow (τ4) components have a higher weight in the decay process, which means that the persistence process is prolonged.
An important remark is that CAO: Eu, Nd white powders are obtained under indoor illumination following the molten salt procedure in comparison with the dark-gray color characteristic of these long-lasting materials. The origin of the dark-gray color, instead of white, in our material and in other oxides synthesized under the N
2/H
2 atmosphere, is attributed to the presence of oxygen vacancies in the structure [
23]. (Left image in
Figure 4c). The right image (
Figure 4c) exhibits the response of the CAO: Eu, Nd powders in darkness after cutting of the excitation.
3.4. Tuning the Functional Properties by Heat Treatment and Eu2+ Content
It is important to evaluate the intensity of the emission and a way to modulate it. As was previously shown, not only the crystallinity and the particle size are the parameters influencing the brightness. The fraction of the Eu
2+ plays a key role in achieving higher intensities [
23]. However, it is far from obvious to determine the fraction of both species from PL spectroscopy [
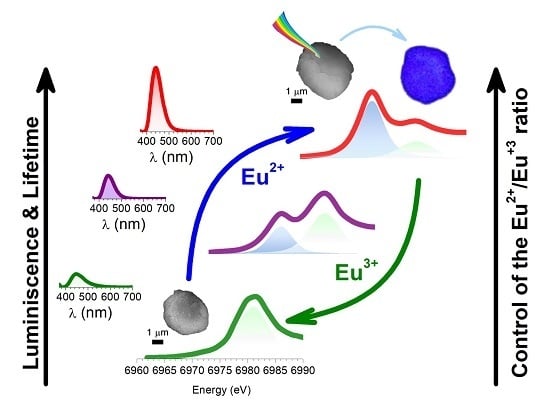
23]. To do that, the XANES technique was employed, which can be considered as a powerful tool to investigate the valence states of the rare-earth dopants, i.e., europium. The significant difference in the absorption energy of the two oxidation states can serve as a signature (6971.8 eV for Eu
2+ and 6979.5 eV for Eu
3+).
As
Figure 5a depicts, an increase in the thermal temperature results in the domination of the absorption peak of the divalent Eu cations. It is worth it to remark that a considerable fraction of Eu
3+ can be detected in the particles of CAO, but the concentration is not reflected by the PL spectrum.
To determine the concentration ratio of Eu
2+ and Eu
3+ in the CAO: Eu, Nd particles, two methods were assessed. Several studies quantify the relative abundance of Eu
2+ and Eu
3+ ions in the samples by means of a linear combination fitting of standards at the Eu L
3-edge absorption. In this case, an absorption signal combination of Eu
2O
3 and EuI
2 standards is performed using the Athena software. The fitting values for Eu L
3-edge XANES spectra analyzed by a linear combination of Eu
2O
3 and EuI
2 references are compiled in
Table 3. However, these analyses do not consider the transition probabilities of the two states (2
+ and 3
+).
Previous studies suggest that the peak amplitude related to Eu
2+ L
3-edge resonance is 1.5 times smaller than that corresponding to the Eu
3+ resonance peak [
24,
25]. Taking this into account, the deconvoluted peak fitting of the white line (WL) at the Eu L
3-edge was done by using a pseudo-Voigt and an arctangent step function for each peak of the absorption spectra [
25,
26] and employing the mole ratio M
Eu through M
Eu = RA
Eu [
25,
27]. The R-value is 1.5, and the A
Eu represents the ratio between the Eu
2+ and Eu
3+ WLs.
Figure 5b shows the best fitting done by modeling the experimental data, and the results taking the reported transition probability difference are summarized in
Table 4. It should be noted that during the XAS experiments, no modifications in the average valence of the samples were identified by the X-ray irradiation.
Despite the fact that from the linear combination fitting, the resulting Eu2+ percentage for the sample thermally treated at 1000 °C shows that Eu2+ is not present, the results obtained by the deconvoluted peak fitting and by PL spectroscopy reflect a low content of Eu2+. It means that in spite of XANES analysis exhibiting an insignificant fraction of Eu2+ cations, this quantity is enough to allow the blue emission of powder. In any case, further research is required to optimize the reduction process via the molten salt route, in order to increase the emission intensity. For samples prepared at 1200 °C and 1400 °C, the Eu2+: Eu3+ fraction is calculated to be around 0.5:0.5 and 1:0, respectively, by both approaches, which agrees with the PL response.
Figure 5b exhibits the percentage of PL against the fraction of Eu
2+ calculated from XANES spectra fitting by the deconvoluted peak and linear combination method. The relative amounts of Eu
2+ determined by two fitting approaches show that the % of PL, calculated for each sample, taking as 100% the PL intensity obtained for the sample sintered (frittage) at 1400 °C, increases when the Eu
2+ fraction increases, following a linear trend. Even so, the tendency is similar in both XANES fitting. The XANES results given from the fitting at the whiteline (WL), considering the transition probabilities of the two Eu
2+ and Eu
3+ states by the deconvoluted peak fitting or by the linear fitting of absorption signal with the Eu
2O
3 and EuI
2 standards, provide the relative abundance of Eu
2+ and Eu
3+. This quantification cannot be assessed by taking into account only the results obtained by PL spectroscopy, showing that the joint characterization by both XANES and PL is necessary to evaluate and optimize the reduction of Eu species.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}