Plasmonic Interferometers as TREM2 Sensors for Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. TREM2 as Biomarker for Early-Onset Detection of AD
1.2. Plasmonic Interferometry for Sensing Applications
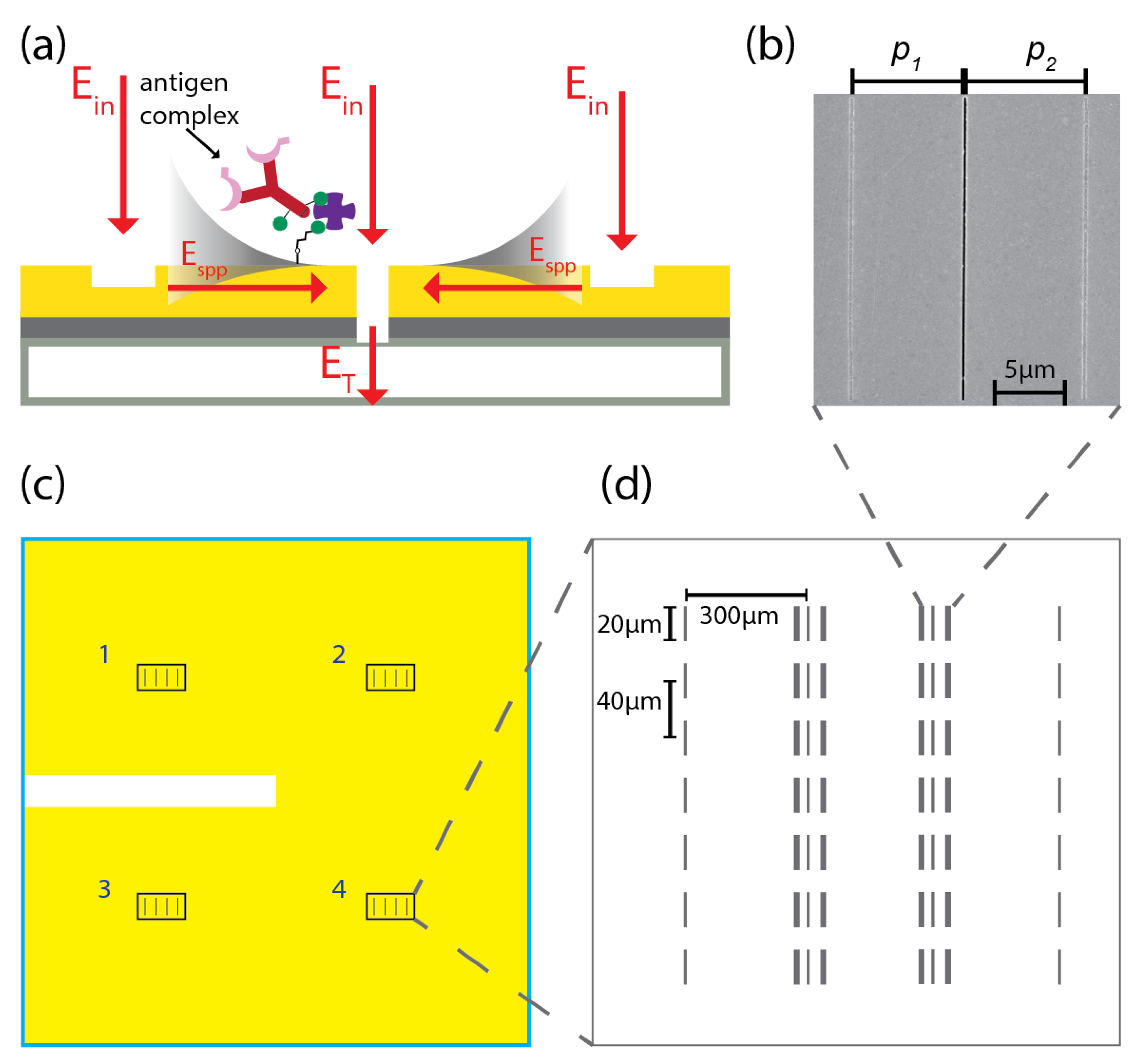
2. Biosensing Chip: Design, Fabrication and Implementation
2.1. Fabrication of Biosensing Chip Based on Plasmonic Interferometry
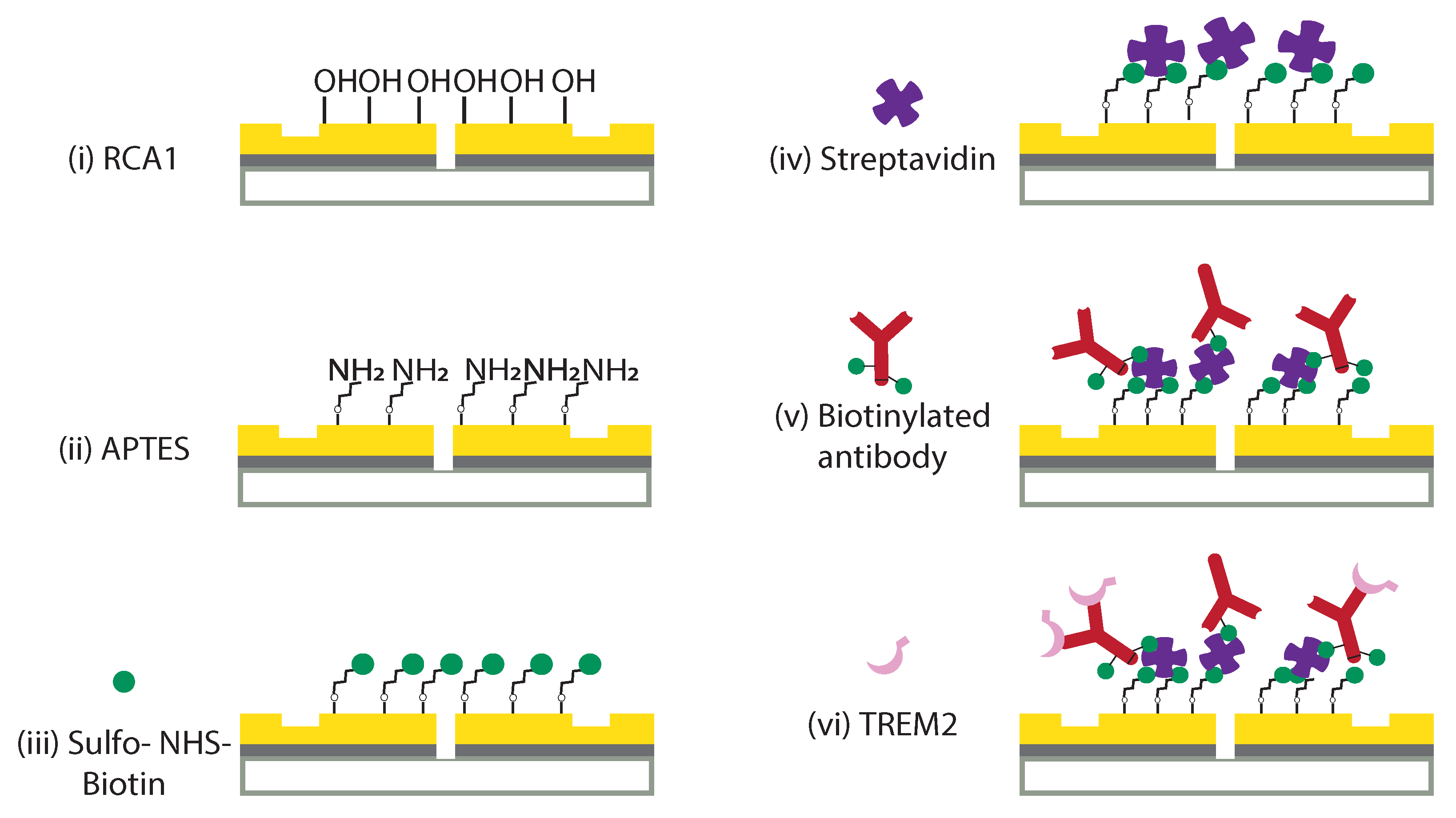
2.2. Surface Functionalization of Optical Biochip
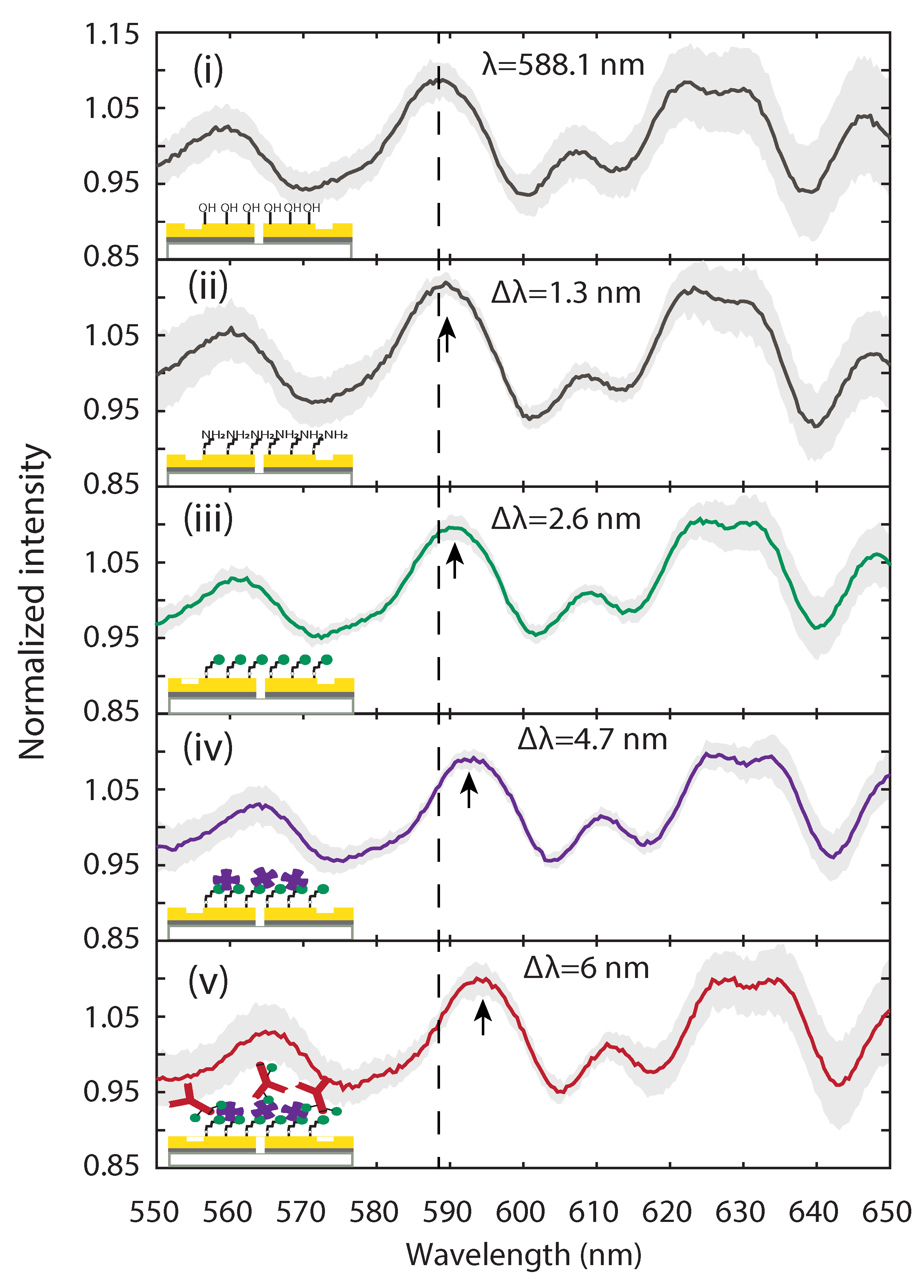
2.3. Optical Characterization of Surface Functionalization Steps with Plasmonic Interferometry
3. TREM2 Biosensing Experiment: Results and Analysis
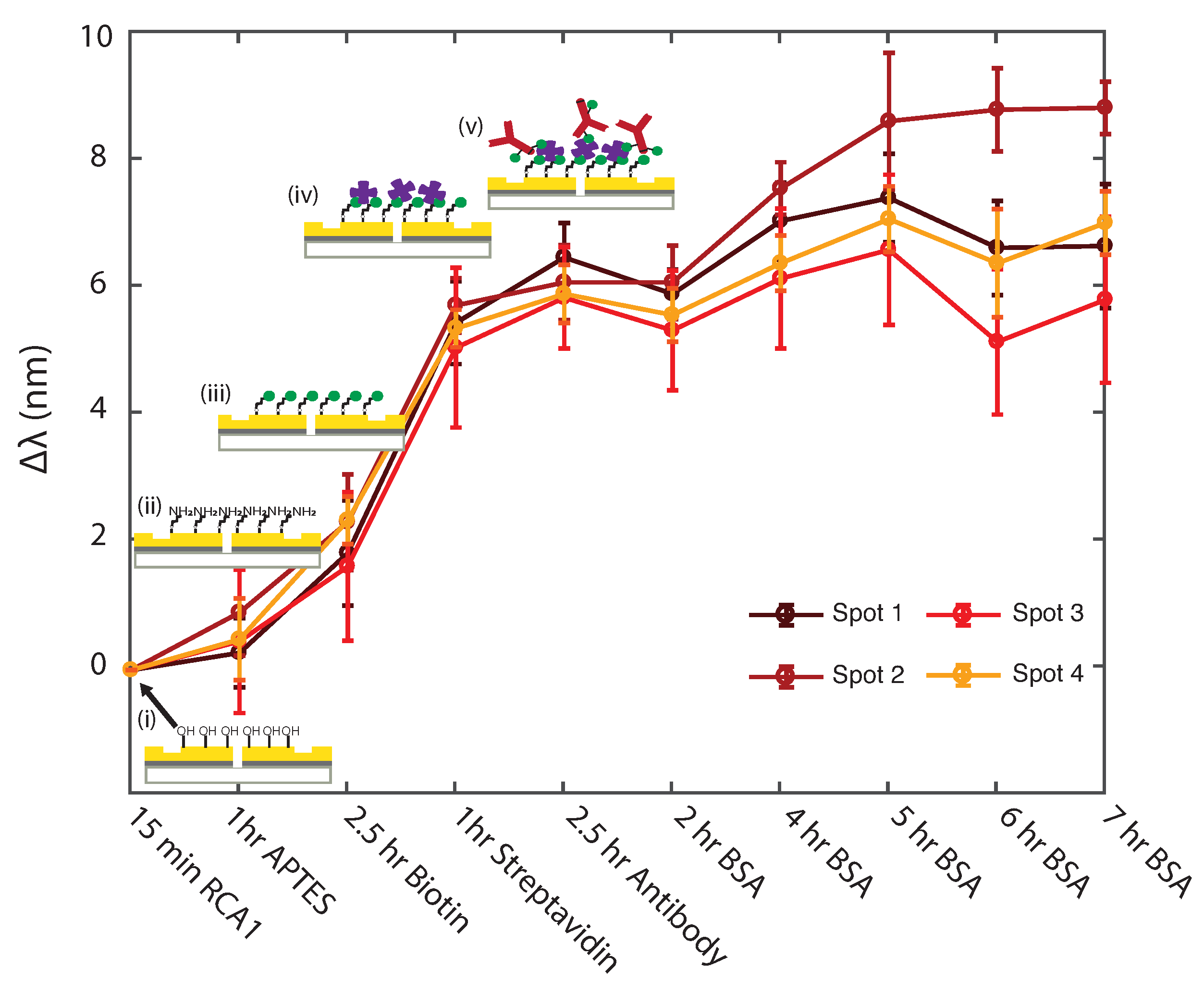
3.1. Uniformity Study of Surface Functionalization Steps with Plasmonic Interferometry
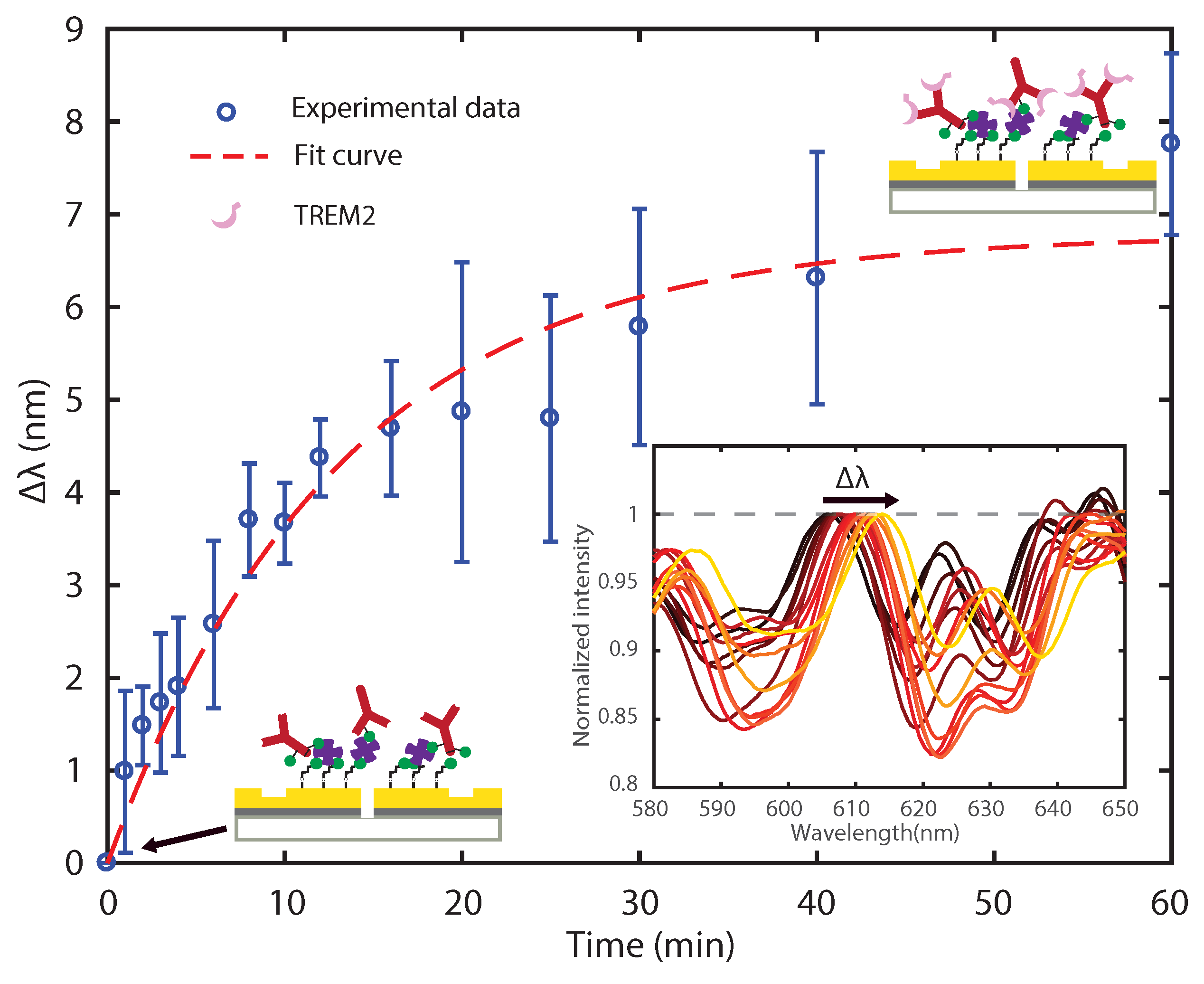
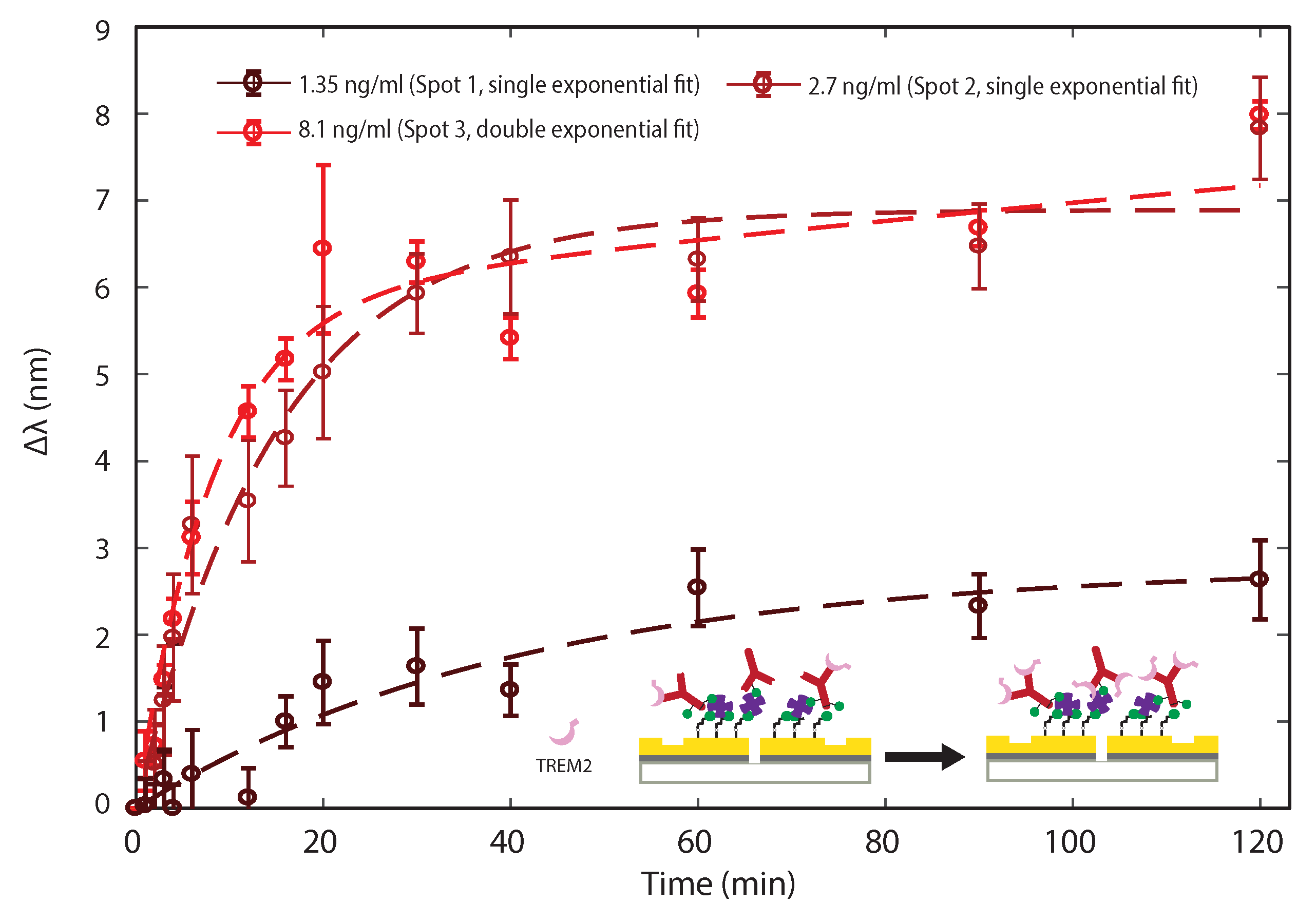
3.2. Sensing TREM2 Antigen-Antibody Binding Interaction with Plasmonic Interferometry
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APTES | (3-Aminopropyl)triethoxysilane |
| BSA | Bovine serum albumin |
| CCD | Charged-coupled device |
| CSF | Cerebrospinal fluid |
| DI | Deionized |
| ELISA | Enzyme-linked immunosorbent assay |
| FIB | Focused ion beam |
| GSG | Groove-Slit-Groove |
| NA | Numerical aperture |
| NHS | N-Hydroxysulfosuccinimide |
| PBS | Phosphate buffer solution |
| RCA1 | Radio Corporation of America Si wafer cleaning procedure, standard clean-1 |
| SEM | Scanning Electron Micrograph/Microscopy |
| SPP | Surface Plasmon Polariton |
| SPR | Surface Plasmon Resonance |
| TREM2 | Triggering Receptor Expressed on Myeloid Cells 2 |
References
- Vos, T.; Allen, C.; Arora, M.; Barber, R.M.; Bhutta, Z.A.; Brown, A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Towards a Dementia Plan: A WHO Guide; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Alzheimer’s Association. 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Calcoen, D.; Elias, L.; Yu, X. What does it take to produce a breakthrough drug? Nat. Rev. Drug Discov. 2015, 14, 161–162. [Google Scholar] [CrossRef]
- Morris, J.C.; McKeel, D.W., Jr.; Fulling, K.; Torack, R.M.; Berg, L. Validation of clinical diagnostic criteria for Alzheimer’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1988, 24, 17–22. [Google Scholar] [CrossRef]
- Plenge, R.M. Disciplined approach to drug discovery and early development. Sci. Transl. Med. 2016, 8, 349ps15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escott-Price, V.; Shoai, M.; Pither, R.; Williams, J.; Hardy, J. Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol. Aging 2017, 49, 214.e7–214.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef]
- Malik, M.; Parikh, I.; Vasquez, J.B.; Smith, C.; Tai, L.; Bu, G.; LaDu, M.J.; Fardo, D.W.; Rebeck, G.W.; Estus, S. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 2015, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, microglia, and neurodegenerative diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef]
- Suárez-Calvet, M.; Kleinberger, G.; Caballero, M.Á.A.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleó, A.; Blesa, R.; Gispert, J.D.; et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016, e201506123. [Google Scholar] [CrossRef]
- Piccio, L.; Deming, Y.; Del-Águila, J.L.; Ghezzi, L.; Holtzman, D.M.; Fagan, A.M.; Fenoglio, C.; Galimberti, D.; Borroni, B.; Cruchaga, C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016, 131, 925–933. [Google Scholar] [CrossRef]
- Pimenova, A.A.; Marcora, E.; Goate, A.M. A tale of two genes: Microglial Apoe and Trem2. Immunity 2017, 47, 398–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kober, D.L.; Alexander-Brett, J.M.; Karch, C.M.; Cruchaga, C.; Colonna, M.; Holtzman, M.J.; Brett, T.J. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife 2016, 5. [Google Scholar] [CrossRef]
- Suárez-Calvet, M.; Caballero, M.Á.A.; Kleinberger, G.; Bateman, R.J.; Fagan, A.M.; Morris, J.C.; Levin, J.; Danek, A.; Ewers, M.; Haass, C.; et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med. 2016, 8, 369ra178. [Google Scholar] [CrossRef] [Green Version]
- Gispert, J.D.; Suárez-Calvet, M.; Monté, G.C.; Tucholka, A.; Falcon, C.; Rojas, S.; Rami, L.; Sánchez-Valle, R.; Lladó, A.; Kleinberger, G.; et al. Cerebrospinal fluid sTREM2 levels are associated with gray matter volume increases and reduced diffusivity in early Alzheimer’s disease. Alzheimer Dementia J. Alzheimer Assoc. 2016, 12, 1259–1272. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratuze, M.; Leyns, C.E.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Jay, T.R.; von Saucken, V.E.; Landreth, G.E. TREM2 in neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, A.; Geddes, T.; Han, B.; Bahado-Singh, R.O.; Wilson, G.D.; Imam, K.; Maddens, M.; Graham, S.F. Diagnostic biomarkers of Alzheimer’s disease as identified in saliva using 1H nmR-based metabolomics. J. Alzheimer Dis. 2017, 58, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Öhrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, E.N.; Swarovski, M.S.; Linortner, P.; Shahid, M.; Zuckerman, A.J.; Wang, Q.; Channappa, D.; Minhas, P.S.; Mhatre, S.D.; Plowey, E.D.; et al. Soluble TREM2 is elevated in Parkinson’s disease subgroups with increased CSF tau. Brain 2020, 143, 932–943. [Google Scholar] [CrossRef]
- Piccio, L.; Buonsanti, C.; Cella, M.; Tassi, I.; Schmidt, R.E.; Fenoglio, C.; Rinker, J.; Naismith, R.T.; Panina-Bordignon, P.; Passini, N.; et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 2008, 131, 3081–3091. [Google Scholar] [CrossRef] [Green Version]
- Haun, J.B.; Yoon, T.J.; Lee, H.; Weissleder, R. Magnetic nanoparticle biosensors. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010, 2, 291–304. [Google Scholar] [CrossRef]
- Vamvakaki, V.; Chaniotakis, N.A. Carbon nanostructures as transducers in biosensors. Sens. Actuators B Chem. 2007, 126, 193–197. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Yeh, H.C.; Kuroki, M.T.; Wang, T.H. Single-quantum-dot-based DNA nanosensor. Nat. Mater. 2005, 4, 826–831. [Google Scholar] [CrossRef]
- Li, Y.; Schluesener, H.J.; Xu, S. Gold nanoparticle-based biosensors. Gold Bull. 2010, 43, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Siu, V.S.; Roelke, A.; Mehta, V.; Rhieu, S.Y.; Palmore, G.T.R.; Pacifici, D. Nanoscale plasmonic interferometers for multispectral, high-throughput biochemical sensing. Nano Lett. 2012, 12, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Graydon, O. Sensing: Plasmonic interferometry. Nat. Photonics 2012, 6, 139. [Google Scholar] [CrossRef]
- Martín-Becerra, D.; Armelles, G.; González, M.; García-Martín, A. Plasmonic and magnetoplasmonic interferometry for sensing. New J. Phys. 2013, 15, 085021. [Google Scholar] [CrossRef] [Green Version]
- Yeung, K.Y.; Yoon, H.; Andress, W.; West, K.; Pfeiffer, L.; Ham, D. Two-path solid-state interferometry using ultra-subwavelength two-dimensional plasmonic waves. Appl. Phys. Lett. 2013, 102, 021104. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Xin, Z.; Gan, Q.; Cheng, X.; Bartoli, F.J. Plasmonic interferometers for label-free multiplexed sensing. Opt. Express 2013, 21, 5859–5871. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xin, Z.; Zeng, B.; Gan, Q.; Cheng, X.; Bartoli, F.J. Plasmonic interferometric sensor arrays for high-performance label-free biomolecular detection. Lab Chip 2013, 13, 4755–4764. [Google Scholar] [CrossRef]
- Gan, Q.; Gao, Y.; Bartoli, F.J. Vertical plasmonic Mach-Zehnder interferometer for sensitive optical sensing. Opt. Express 2009, 17, 20747–20755. [Google Scholar] [CrossRef]
- Gao, Y.; Gan, Q.; Xin, Z.; Cheng, X.; Bartoli, F.J. Plasmonic Mach–Zehnder interferometer for ultrasensitive on-chip biosensing. ACS Nano 2011, 5, 9836–9844. [Google Scholar] [CrossRef]
- Li, X.; Tan, Q.; Bai, B.; Jin, G. Non-spectroscopic refractometric nanosensor based on a tilted slit-groove plasmonic interferometer. Opt. Express 2011, 19, 20691–20703. [Google Scholar] [CrossRef]
- Siu, V.S.; Feng, J.; Flanigan, P.W.; Palmore, G.T.R.; Pacifici, D. A “plasmonic cuvette”: Dye chemistry coupled to plasmonic interferometry for glucose sensing. Nanophotonics 2014, 3, 125–140. [Google Scholar] [CrossRef]
- Cetin, A.E.; Coskun, A.F.; Galarreta, B.C.; Huang, M.; Herman, D.; Ozcan, A.; Altug, H. Handheld high-throughput plasmonic biosensor using computational on-chip imaging. Light. Sci. Appl. 2014, 3, e122. [Google Scholar] [CrossRef]
- Sun, X.; Dai, D.; Thylén, L.; Wosinski, L. High-sensitivity liquid refractive-index sensor based on a Mach-Zehnder interferometer with a double-slot hybrid plasmonic waveguide. Opt. Express 2015, 23, 25688–25699. [Google Scholar] [CrossRef]
- Li, D.; Feng, J.; Pacifici, D. Nanoscale optical interferometry with incoherent light. Sci. Rep. 2016, 6, 20836. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Feng, J.; Pacifici, D. Higher-order surface plasmon contributions to passive and active plasmonic interferometry. Opt. Express 2016, 24, 27309–27318. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, D.; Pacifici, D. Circular slit-groove plasmonic interferometers: A generalized approach to high-throughput biochemical sensing. Opt. Mater. Express 2015, 5, 2742–2753. [Google Scholar] [CrossRef]
- Raether, H. Surface plasmons on smooth surfaces. In Surface Plasmons on Smooth and Rough Surfaces and on Gratings; Springer: Berlin, Germany, 1988; pp. 4–39. [Google Scholar]
- Feng, J.; Pacifici, D. A spectroscopic refractometer based on plasmonic interferometry. J. Appl. Phys. 2016, 119, 083104. [Google Scholar] [CrossRef] [Green Version]
- Morrill, D.; Li, D.; Pacifici, D. Measuring subwavelength spatial coherence with plasmonic interferometry. Nat. Photonics 2016, 10, 681–687. [Google Scholar] [CrossRef]
- Vashist, S.K.; Dixit, C.K.; MacCraith, B.D.; O’Kennedy, R. Effect of antibody immobilization strategies on the analytical performance of a surface plasmon resonance-based immunoassay. Analyst 2011, 136, 4431. [Google Scholar] [CrossRef]
- Williams, E.H.; Davydov, A.V.; Motayed, A.; Sundaresan, S.G.; Bocchini, P.; Richter, L.J.; Stan, G.; Steffens, K.; Zangmeister, R.; Schreifels, J.A.; et al. Immobilization of streptavidin on 4H–SiC for biosensor development. Appl. Surf. Sci. 2012, 258, 6056–6063. [Google Scholar] [CrossRef]
- Xiao, Y.; Isaacs, S.N. Enzyme-linked immunosorbent assay (ELISA) and blocking with bovine serum albumin (BSA)—not all BSAs are alike. J. Immunol. Methods 2012, 384, 148–151. [Google Scholar] [CrossRef] [Green Version]
- Malmqvist, M. Surface plasmon resonance for detection and measurement of antibody-antigen affinity and kinetics. Curr. Opin. Immunol. 1993, 5, 282–286. [Google Scholar] [CrossRef]
- O’Shannessy, D.J.; Brigham-Burke, M.; Soneson, K.K.; Hensley, P.; Brooks, I. Determination of rate and equilibrium binding constants for macromolecular interactions using surface plasmon resonance: Use of nonlinear least squares analysis methods. Anal. Biochem. 1993, 212, 457–468. [Google Scholar] [CrossRef]
- O’Shannessy, D.J.; Brigham-Burke, M.; Soneson, K.K.; Hensley, P.; Brooks, I. Determination of rate and equilibrium binding constants for macromolecular interactions by surface plasmon resonance. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1994; Volume 240, pp. 323–349. [Google Scholar]
- O’Shannessy, D.J. Determination of kinetic rate and equilibrium binding constants for macromolecular interactions: A critique of the surface plasmon resonance literature. Curr. Opin. Biotechnol. 1994, 5, 65–71. [Google Scholar] [CrossRef]
- Myszka, D.G. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Curr. Opin. Biotechnol. 1997, 8, 50–57. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Teichroeb, J.; Forrest, J.; Jones, L.; Chan, J.; Dalton, K. Quartz crystal microbalance study of protein adsorption kinetics on poly(2-hydroxyethyl methacrylate). J. Colloid Interface Sci. 2008, 325, 157–164. [Google Scholar] [CrossRef]
- Holmberg, M.; Hou, X. Competitive Protein Adsorption—Multilayer Adsorption and Surface Induced Protein Aggregation. Langmuir 2009, 25, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Ball, V.; Ramsden, J.J. Analysis of hen egg white lysozyme adsorption on Si (Ti) O2 aqueous solution interfaces at low ionic strength: A biphasic reaction related to solution self-association. Colloids Surfaces B Biointerfaces 2000, 17, 81–94. [Google Scholar] [CrossRef]
- Haynes, C.A.; Norde, W. Globular proteins at solid/liquid interfaces. Colloids Surfaces B Biointerfaces 1994, 2, 517–566. [Google Scholar] [CrossRef]
- Rabe, M.; Verdes, D.; Zimmermann, J.; Seeger, S. Surface Organization and Cooperativity during Nonspecific Protein Adsorption Events. J. Phys. Chem. B 2008, 112, 13971–13980. [Google Scholar] [CrossRef] [PubMed]
- Minton, A.P. Effects of excluded surface area and adsorbate clustering on surface adsorption of proteins: I. Equilibrium models. Biophys. Chem. 2000, 86, 239–247. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Odessey, R.; Li, D.; Pacifici, D. Plasmonic Interferometers as TREM2 Sensors for Alzheimer’s Disease. Biosensors 2021, 11, 217. https://doi.org/10.3390/bios11070217
Li D, Odessey R, Li D, Pacifici D. Plasmonic Interferometers as TREM2 Sensors for Alzheimer’s Disease. Biosensors. 2021; 11(7):217. https://doi.org/10.3390/bios11070217
Chicago/Turabian StyleLi, Dingdong, Rachel Odessey, Dongfang Li, and Domenico Pacifici. 2021. "Plasmonic Interferometers as TREM2 Sensors for Alzheimer’s Disease" Biosensors 11, no. 7: 217. https://doi.org/10.3390/bios11070217
APA StyleLi, D., Odessey, R., Li, D., & Pacifici, D. (2021). Plasmonic Interferometers as TREM2 Sensors for Alzheimer’s Disease. Biosensors, 11(7), 217. https://doi.org/10.3390/bios11070217