Spatial Omics Sequencing Based on Microfluidic Array Chips



Abstract
:1. Introduction
2. General Classification of Spatial Profiling Technology
3. Spatial Transcriptomics Sequencing Methods Based on Barcoded Array Chips
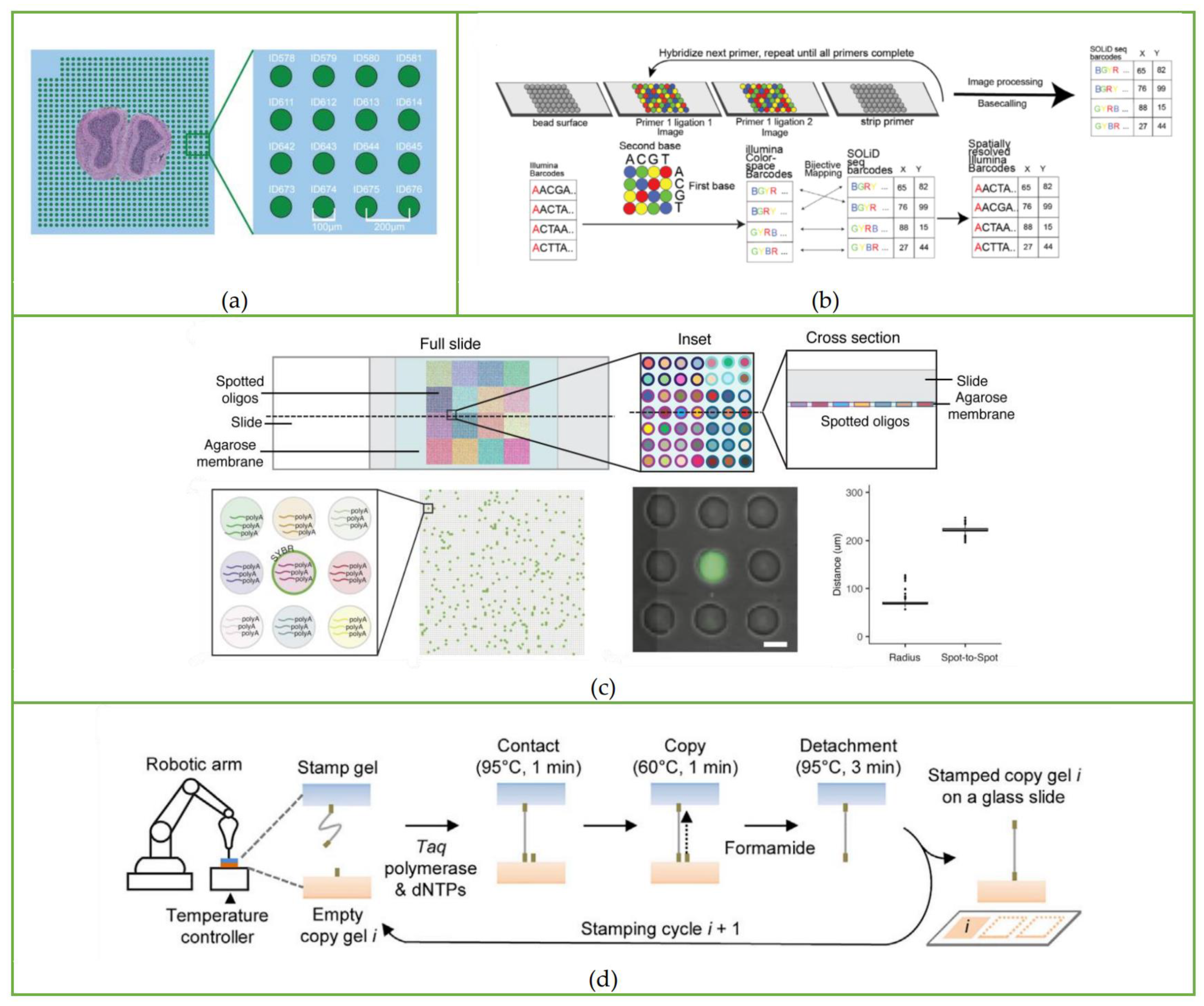
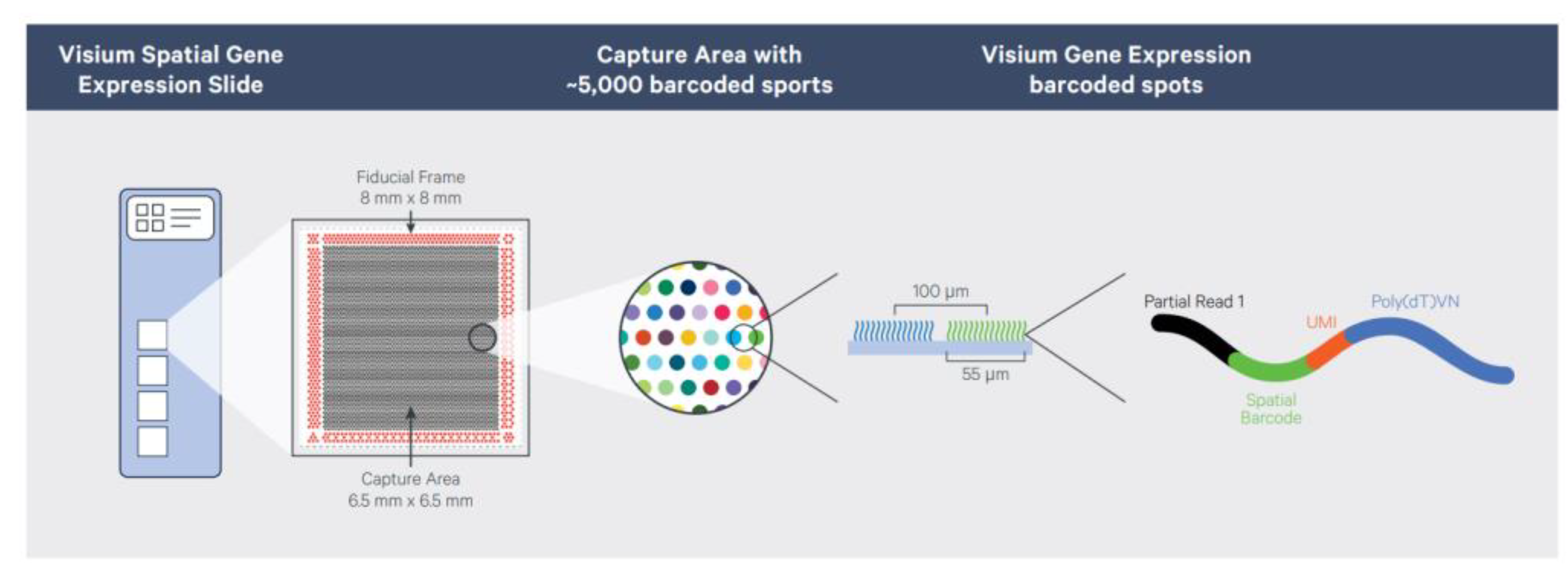
3.1. ST Method
3.2. Slide-Seq
3.3. HDST Method
3.4. Seq-Scope
3.5. Sci-Space
3.6. Stereo-Seq
3.7. Pixel-Seq
4. Spatial Multi-Omic Sequencing Methods Based on Microfluidic Chips
4.1. DBiT-Seq
4.2. Spatial-CUT&Tag
4.3. Spatial-ATAC-Seq Strategy
4.4. Spatial-CITE-Seq
4.5. Stereo-CITE-Seq
5. Commercialization of Spatial Sequencing Methods
6. Discussion and Future Perspectives
6.1. Limitations and Possible Improvements of Barcoded Array Chips
6.2. Bridging the Gap between Single-Cell Sequencing and Spatial Omics Sequencing
6.3. Multi-Omic Sequencing Apply in Spatial Omics Sequencing
6.4. The Needs of Real Three-Dimensional Spatial Profiling
6.5. Limitations in the Application of Spatial Omics Sequencing and Possible Solutions
6.6. The Need for More Advanced Commercial Spatial Profiling Technology
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baron, C.S.; van Oudenaarden, A. Unravelling cellular relationships during development and regeneration using genetic lineage tracing. Nat. Rev. Mol. Cell Biol. 2019, 20, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Cui, G.; Ke, J.; Jing, N. Using Single-Cell and Spatial Transcriptomes to Understand Stem Cell Lineage Specification During Early Embryo Development. Annu. Rev. Genom. Hum. Genet. 2020, 21, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; Mignardi, M.; Pacureanu, A.; Svedlund, J.; Botling, J.; Wählby, C.; Nilsson, M. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 2013, 10, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstråhle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.; Larsson, L.; et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. [Google Scholar] [CrossRef] [PubMed]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514. [Google Scholar] [CrossRef]
- Codeluppi, S.; Borm, L.E.; Zeisel, A.; La Manno, G.; van Lunteren, J.A.; Svensson, C.I.; Linnarsson, S. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat. Methods 2018, 15, 932–935. [Google Scholar] [CrossRef]
- Chen, W.T.; Lu, A.; Craessaerts, K.; Pavie, B.; Sala Frigerio, C.; Corthout, N.; Qian, X.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991. [Google Scholar] [CrossRef]
- Maynard, K.R.; Collado-Torres, L.; Weber, L.M.; Uytingco, C.; Barry, B.K.; Williams, S.R.; Catallini, J.L., 2nd; Tran, M.N.; Besich, Z.; Tippani, M.; et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci. 2021, 24, 425–436. [Google Scholar] [CrossRef]
- Desai, N.; Neyaz, A.; Szabolcs, A.; Shih, A.R.; Chen, J.H.; Thapar, V.; Nieman, L.T.; Solovyov, A.; Mehta, A.; Lieb, D.J.; et al. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat. Commun. 2020, 11, 6319. [Google Scholar] [CrossRef]
- Boyd, D.F.; Allen, E.K.; Randolph, A.G.; Guo, X.J.; Weng, Y.; Sanders, C.J.; Bajracharya, R.; Lee, N.K.; Guy, C.S.; Vogel, P.; et al. Exuberant fibroblast activity compromises lung function via ADAMTS4. Nature 2020, 587, 466–471. [Google Scholar] [CrossRef]
- Giacomello, S.; Salmén, F.; Terebieniec, B.K.; Vickovic, S.; Navarro, J.F.; Alexeyenko, A.; Reimegård, J.; McKee, L.S.; Mannapperuma, C.; Bulone, V.; et al. Spatially resolved transcriptome profiling in model plant species. Nat. Plants 2017, 3, 17061. [Google Scholar] [CrossRef]
- Burgess, D.J. Spatial transcriptomics coming of age. Nat. Rev. Genet. 2019, 20, 317. [Google Scholar] [CrossRef]
- Xiaowei. Method of the Year 2020: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 1. [Google Scholar] [CrossRef]
- Raj, A.; van den Bogaard, P.; Rifkin, S.A.; van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef] [Green Version]
- Lubeck, E.; Cai, L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods 2012, 9, 743–748. [Google Scholar] [CrossRef]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Yang, J.L.; Ferrante, T.C.; Terry, R.; Jeanty, S.S.; Li, C.; Amamoto, R.; et al. Highly multiplexed subcellular RNA sequencing in situ. Science 2014, 343, 1360–1363. [Google Scholar] [CrossRef] [Green Version]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F.; et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef]
- Cho, C.S.; Xi, J.; Si, Y.; Park, S.R.; Hsu, J.E.; Kim, M.; Jun, G.; Kang, H.M.; Lee, J.H. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 2021, 184, 3559–3572. [Google Scholar] [CrossRef]
- Srivatsan, S.R.; Regier, M.C.; Barkan, E.; Franks, J.M.; Packer, J.S.; Grosjean, P.; Duran, M.; Saxton, S.; Ladd, J.J.; Spielmann, M.; et al. Embryo-scale, single-cell spatial transcriptomics. Science 2021, 373, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 2022, 185, 1777–1792. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Sun, L.; Dong, R.; Chen, J.Y.; Silakit, R.; Condon, L.F.; Lin, Y.; Lin, S.; Palmiter, R.D.; Gu, L. Polony gels enable amplifiable DNA stamping and spatial transcriptomics of chronic pain. Cell 2022, 185, 4621–4633. [Google Scholar] [CrossRef] [PubMed]
- Stickels, R.R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.L.; Di Bella, D.J.; Arlotta, P.; Macosko, E.Z.; Chen, F. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 2021, 39, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Packer, J.S.; Ramani, V.; Cusanovich, D.A.; Huynh, C.; Daza, R.; Qiu, X.; Lee, C.; Furlan, S.N.; Steemers, F.J.; et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 2017, 357, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef]
- Srivatsan, S.R.; McFaline-Figueroa, J.L.; Ramani, V.; Saunders, L.; Cao, J.; Packer, J.; Pliner, H.A.; Jackson, D.L.; Daza, R.M.; Christiansen, L.; et al. Massively multiplex chemical transcriptomics at single-cell resolution. Science 2020, 367, 45–51. [Google Scholar] [CrossRef]
- Zhu, C.; Preissl, S.; Ren, B. Single-cell multimodal omics: The power of many. Nat. Methods 2020, 17, 11–14. [Google Scholar] [CrossRef]
- Fessenden, M. Technologies to watch in 2019. Nature 2019, 565, 521–523. [Google Scholar] [CrossRef] [Green Version]
- Teichmann, S.; Efremova, M. Method of the Year 2019: Single-cell multimodal omics. Nat. Methods 2020, 17, 1. [Google Scholar]
- Mulqueen, R.M.; Pokholok, D.; Norberg, S.J.; Torkenczy, K.A.; Fields, A.J.; Sun, D.; Sinnamon, J.R.; Shendure, J.; Trapnell, C.; O’Roak, B.J.; et al. Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol. 2018, 36, 428–431. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Telenius, H.; Carter, N.P.; Bebb, C.E.; Nordenskjöld, M.; Ponder, B.A.; Tunnacliffe, A. Degenerate oligonucleotide-primed PCR: General amplification of target DNA by a single degenerate primer. Genomics 1992, 13, 718–725. [Google Scholar] [CrossRef]
- Dean, F.B.; Hosono, S.; Fang, L.; Wu, X.; Faruqi, A.F.; Bray-Ward, P.; Sun, Z.; Zong, Q.; Du, Y.; Du, J.; et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA 2002, 99, 5261–5266. [Google Scholar] [CrossRef]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Xing, D.; Tan, L.; Li, H.; Zhou, G.; Huang, L.; Xie, X.S. Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 2017, 356, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Genshaft, A.S.; Li, S.; Gallant, C.J.; Darmanis, S.; Prakadan, S.M.; Ziegler, C.G.; Lundberg, M.; Fredriksson, S.; Hong, J.; Regev, A.; et al. Multiplexed, targeted profiling of single-cell proteomes and transcriptomes in a single reaction. Genome Biol. 2016, 17, 188. [Google Scholar] [CrossRef] [Green Version]
- Peterson, V.M.; Zhang, K.X.; Kumar, N.; Wong, J.; Li, L.; Wilson, D.C.; Moore, R.; McClanahan, T.K.; Sadekova, S.; Klappenbach, J.A. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 2017, 35, 936–939. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665–1681. [Google Scholar] [CrossRef]
- Deng, Y.; Bartosovic, M.; Kukanja, P.; Zhang, D.; Liu, Y.; Su, G.; Enninful, A.; Bai, Z.; Castelo-Branco, G.; Fan, R. Spatial-CUT&Tag: Spatially resolved chromatin modification profiling at the cellular level. Science 2022, 375, 681–686. [Google Scholar]
- Deng, Y.; Bartosovic, M.; Ma, S.; Zhang, D.; Kukanja, P.; Xiao, Y.; Su, G.; Liu, Y.; Qin, X.; Rosoklija, G.B.; et al. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature 2022, 609, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; DiStasio, M.; Su, G.; Asashima, H.; Enninful, A.; Qin, X.; Deng, Y.; Nam, J.; Gao, F.; Bordignon, P.; et al. High-plex protein and whole transcriptome co-mapping at cellular resolution with spatial CITE-seq. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Heng, Y.; Liu, W.; Xiang, J.; Ma, Y.; Chen, L.; Feng, X.; Jia, D.; Liang, D.; Huang, C.; et al. Integrated Spatial Transcriptomic and Proteomic Analysis of Fresh Frozen Tissue Based on Stereo-seq. bioRxiv 2023. [Google Scholar] [CrossRef]
- Bäckdahl, J.; Franzén, L.; Massier, L.; Li, Q.; Jalkanen, J.; Gao, H.; Andersson, A.; Bhalla, N.; Thorell, A.; Rydén, M.; et al. Spatial mapping reveals human adipocyte subpopulations with distinct sensitivities to insulin. Cell Metab. 2021, 33, 1869–1882. [Google Scholar] [CrossRef]
- Hasel, P.; Rose, I.V.L.; Sadick, J.S.; Kim, R.D.; Liddelow, S.A. Neuroinflammatory astrocyte subtypes in the mouse brain. Nat. Neurosci. 2021, 24, 1475–1487. [Google Scholar] [CrossRef]
- Wu, R.; Guo, W.; Qiu, X.; Wang, S.; Sui, C.; Lian, Q.; Wu, J.; Shan, Y.; Yang, Z.; Yang, S.; et al. Comprehensive analysis of spatial architecture in primary liver cancer. Sci. Adv. 2021, 7, eabg3750. [Google Scholar] [CrossRef]
- Meylan, M.; Petitprez, F.; Becht, E.; Bougoüin, A.; Pupier, G.; Calvez, A.; Giglioli, I.; Verkarre, V.; Lacroix, G.; Verneau, J.; et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 2022, 55, 527–541. [Google Scholar] [CrossRef]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, S.; Yao, M.; Chao, N.; Yang, Y.; Song, T.; Ni, Y.; Liu, Z.; Li, W. Spatial transcriptome sequencing revealed spatial trajectory in the Non-Small Cell Lung Carcinoma. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gao, S.; Shi, Q.; Zhang, Y.; Liang, G.; Kang, Z.; Huang, B.; Ma, D.; Wang, L.; Jiao, J.; Fang, X.; et al. Identification of HSC/MPP expansion units in fetal liver by single-cell spatiotemporal transcriptomics. Cell Res. 2022, 32, 38–53. [Google Scholar] [CrossRef]
- Sun, J.; Yuan, Y.; Wu, X.; Liu, A.; Wang, J.; Yang, S.; Liu, B.; Kong, Y.; Wang, L.; Zhang, K.; et al. Excitatory SST neurons in the medial paralemniscal nucleus control repetitive self-grooming and encode reward. Neuron 2022, 110, 3356–3373. [Google Scholar] [CrossRef]
- Gouin, K.H., 3rd; Ing, N.; Plummer, J.T.; Rosser, C.J.; Be, C.B.; Oh, C.; Chen, S.S.; Chan, K.S.; Furuya, H.; Tourtellotte, W.G.; et al. An N-Cadherin 2 expressing epithelial cell subpopulation predicts response to surgery, chemotherapy and immunotherapy in bladder cancer. Nat. Commun. 2021, 12, 4906. [Google Scholar] [CrossRef]
- Porritt, R.A.; Zemmour, D.; Abe, M.; Lee, Y.; Narayanan, M.; Carvalho, T.T.; Gomez, A.C.; Martinon, D.; Santiskulvong, C.; Fishbein, M.C.; et al. NLRP3 Inflammasome Mediates Immune-Stromal Interactions in Vasculitis. Circ. Res. 2021, 129, e183–e200. [Google Scholar] [CrossRef]
- Novel Insights about Your Tissue. Visualized. Spatial Transcriptomics—10× Genomics. Available online: https://www.10xgenomics.com/resources/document-library/756a6a (accessed on 4 May 2023).
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef]
- Toki, M.I.; Merritt, C.R.; Wong, P.F.; Smithy, J.W.; Kluger, H.M.; Syrigos, K.N.; Ong, G.T.; Warren, S.E.; Beechem, J.M.; Rimm, D.L. High-Plex Predictive Marker Discovery for Melanoma Immunotherapy-Treated Patients Using Digital Spatial Profiling. Clin. Cancer Res. 2019, 25, 5503–5512. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, P.; Wang, B.G.; Murdock, T.; Cope, L.; Hsu, F.C.; Wang, T.L.; Shih, I.M. Spatial Transcriptomic Analysis of Ovarian Cancer Precursors Reveals Reactivation of IGFBP2 during Pathogenesis. Cancer Res. 2022, 82, 4528–4541. [Google Scholar] [CrossRef]
- Kumar, V.; Randhawa, P.; Bilodeau, R.; Mercola, D.; McClelland, M.; Agrawal, A.; Nguyen, J.; Castro, P.; Ittmann, M.M.; Rahmatpanah, F. Spatial Profiling of the Prostate Cancer Tumor Microenvironment Reveals Multiple Differences in Gene Expression and Correlation with Recurrence Risk. Cancers 2022, 14, 4923. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Johnson, D.B.; McDonnell, W.J.; Gonzalez-Ericsson, P.I.; Al-Rohil, R.N.; Mobley, B.C.; Salem, J.E.; Wang, D.Y.; Sanchez, V.; Wang, Y.; Chastain, C.A.; et al. A case report of clonal EBV-like memory CD4+T cell activation in fatal checkpoint inhibitor-induced encephalitis. Nat. Med. 2019, 25, 1243–1250. [Google Scholar] [CrossRef]
- Noll, J.M.; Augello, C.J.; Kürüm, E.; Pan, L.; Pavenko, A.; Nam, A.; Ford, B.D. Spatial Analysis of Neural Cell Proteomic Profiles Following Ischemic Stroke in Mice Using High-Plex Digital Spatial Profiling. Mol. Neurobiol. 2022, 59, 7236–7252. [Google Scholar] [CrossRef]
- Rendeiro, A.F.; Ravichandran, H.; Bram, Y.; Chandar, V.; Kim, J.; Meydan, C.; Park, J.; Foox, J.; Hether, T.; Warren, S.; et al. The spatial landscape of lung pathology during COVID-19 progression. Nature 2021, 593, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, C.M.; Cha, J.H.; Park, J.Y.; Yu, Y.S.; Wang, H.J.; Sung, P.S.; Jung, E.S.; Bae, S.H. Multiplexed Digital Spatial Protein Profiling Reveals Distinct Phenotypes of Mononuclear Phagocytes in Livers with Advanced Fibrosis. Cells 2022, 11, 3387. [Google Scholar] [CrossRef] [PubMed]
- Wilbrey-Clark, A.; Roberts, K.; Teichmann, S.A. Cell Atlas technologies and insights into tissue architecture. Biochem. J. 2020, 477, 1427–1442. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, I.C.; Haerty, W.; Kumar, P.; Li, Y.I.; Hu, T.X.; Teng, M.J.; Goolam, M.; Saurat, N.; Coupland, P.; Shirley, L.M.; et al. G&T-seq: Parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods 2015, 12, 519–522. [Google Scholar]
- Zhu, C.; Yu, M.; Huang, H.; Juric, I.; Abnousi, A.; Hu, R.; Lucero, J.; Behrens, M.M.; Hu, M.; Ren, B. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat. Struct. Mol. Biol. 2019, 26, 1063–1070. [Google Scholar]
- Zhu, C.; Zhang, Y.; Li, Y.E.; Lucero, J.; Behrens, M.M.; Ren, B. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat. Methods 2021, 18, 283–292. [Google Scholar] [CrossRef]
- Angermueller, C.; Clark, S.J.; Lee, H.J.; Macaulay, I.C.; Teng, M.J.; Hu, T.X.; Krueger, F.; Smallwood, S.; Ponting, C.P.; Voet, T.; et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 2016, 13, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yuan, P.; Yan, Z.; Yang, M.; Huo, Y.; Nie, Y.; Zhu, X.; Qiao, J.; Yan, L. Single-cell multiomics sequencing reveals the functional regulatory landscape of early embryos. Nat. Commun. 2021, 12, 1247. [Google Scholar] [CrossRef]
- Bergenstråhle, J.; Larsson, L.; Lundeberg, J. Seamless integration of image and molecular analysis for spatial transcriptomics workflows. BMC Genom. 2020, 21, 482. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Publication Year | Omics Involved | Resolution | Single-Cell Level | Detection Area | Cost | Reference |
|---|---|---|---|---|---|---|---|
| ST method | 2016 | Transcriptomics | Spot diameter: 100 μm Center-to-center: 200 μm | Not single-cell level | 6.2 mm × 6.6 mm | Not mentioned | Ståhl P.L., et al. Nat. Methods, 2016 [17] |
| Slide-seq | 2019 | Transcriptomics | Bead diameter: 10 μm | Nearly single-cell level | 3 mm diameter puck | ~$0.10 chip fabrication and ~$200–500 sequencing/3 mm puck | Rodriques S.G., et al. Science, 2019 [18] |
| HDST method | 2019 | Transcriptomics | Bead diameter: 2 μm Well diameter: 2.05 μm | Nearly single-cell level | 5.7 mm × 2.4 mm | Not mentioned | Vickovic S., et al. Nat. Methods, 2019 [19] |
| Seq-Scope | 2021 | Transcriptomics | Center-to-center: ~0.6 μm | Single-cell/subcellular level | 10 mm × 10 mm | Total ~$150/mm2 | Cho C.S., et al. Cell, 2021 [20] |
| Sci-Space | 2021 | Transcriptomics | Spot radius: ~73 μm Center-to-center: ~200 μm | Single-cell | 18 mm × 18 mm | Not mentioned | Srivatsan S.R., et al. Science, 2021 [21] |
| Stereo-seq | 2022 | Transcriptomics | Spot diameter: 0.22 μm Center-to-center: 0.5/0.715 μm | Single-cell/subcellular level | 13.2 m × 13.2 cm | ~$35 chip fabrication and ~$8 sequencing/mm2 | Chen A., et al. Cell, 2022 [22] |
| Pixel-seq | 2022 | Transcriptomics | Polony diameter: 1 μm | Single-cell level | 7 mm × 7 mm | ~$0.06 chip fabrication and ~$60 sequencing/mm2 | Fu X., et al. Cell, 2022 [23] |
| DBiT-seq | 2020 | Transcriptomics and proteomics | Microchannel width: 10/25/50 μm | Nearly single-cell level | 5 mm × 5 mm for maximum | Not mentioned | Liu Y., et al. Cell, 2020 [39] |
| Spatial-CUT&Tag | 2022 | Transcriptomics and histone modification | Microchannel width: 20/50 μm | Not single-cell level | 5 mm × 5 mm for maximum | Not mentioned | Deng Y., et al. Science, 2022 [40] |
| Spatial-ATAC-seq | 2022 | Transcriptomics and chromatin accessibility | Microchannel width: 20 μm | Not single-cell level | 2 mm × 2 mm | Not mentioned | Deng Y., et al. Nature, 2022 [41] |
| Spatial-CITE-seq | 2023 | Transcriptomics and proteomics | Microchannel width: 25 μm | Single-cell level | 2.5 mm × 2.5 mm | Not mentioned | Liu Y., et al. Nat. Biotechnol, 2023 [42] |
| Stereo-CITE-seq | 2023 | Transcriptomics and proteomics | Spot diameter: 0.22 μm Center-to-center: 0.5/0.715 μm | Single-cell level | 13.2 cm × 13.2 cm | Not mentioned | Liao S, et al. bioRxiv, 2023 [43] |
| 10x Genomics Visium | NanoString GeoMx DSP | |
|---|---|---|
| Tissue compatibility | Embedding frozen samples or FFPE paraffin samples | Any sample, FFPE, or fresh frozen samples |
| Tissue area | 6.5 mm × 6.5 mm | 14.6 mm × 36.2 mm |
| Detection area | Capture area | Regions of interest (ROIs) |
| Resolution | 1–10 cells/spot | >100 cells/ROI |
| RNA capture method | polyA capture or specific probe hybridization | Specific probe hybridization |
| Species limitation | Eukaryote | Human and mouse |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, J.; Pan, Y.; Liu, X.; Cao, W.; Mu, Y.; Zhu, Q. Spatial Omics Sequencing Based on Microfluidic Array Chips. Biosensors 2023, 13, 712. https://doi.org/10.3390/bios13070712
Shi J, Pan Y, Liu X, Cao W, Mu Y, Zhu Q. Spatial Omics Sequencing Based on Microfluidic Array Chips. Biosensors. 2023; 13(7):712. https://doi.org/10.3390/bios13070712
Chicago/Turabian StyleShi, Jianyu, Yating Pan, Xudong Liu, Wenjian Cao, Ying Mu, and Qiangyuan Zhu. 2023. "Spatial Omics Sequencing Based on Microfluidic Array Chips" Biosensors 13, no. 7: 712. https://doi.org/10.3390/bios13070712
APA StyleShi, J., Pan, Y., Liu, X., Cao, W., Mu, Y., & Zhu, Q. (2023). Spatial Omics Sequencing Based on Microfluidic Array Chips. Biosensors, 13(7), 712. https://doi.org/10.3390/bios13070712