

Mutational Signatures as Sensors of Environmental Exposures: Analysis of Smoking-Induced Lung Tissue Remodeling


Abstract
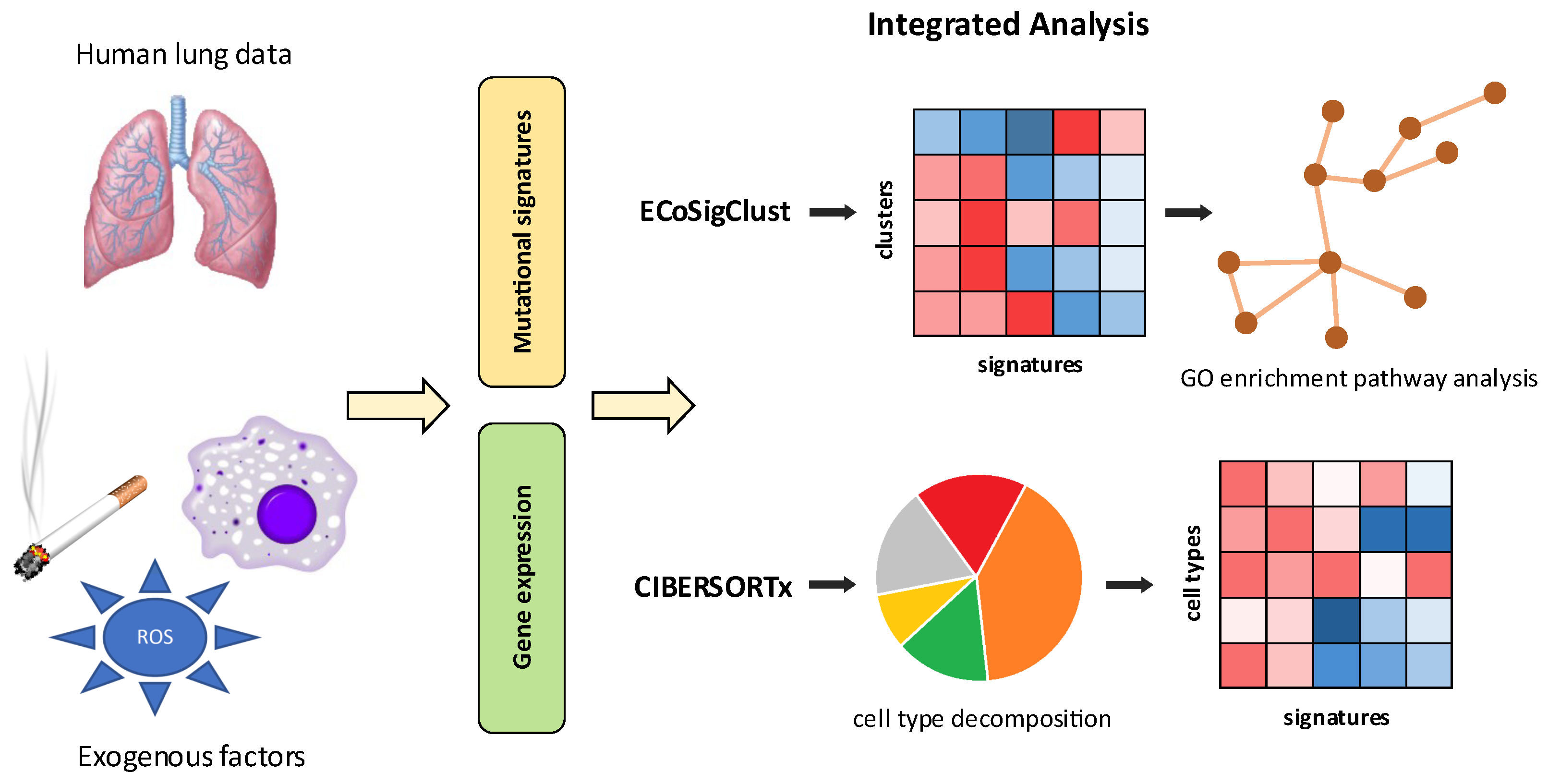
:1. Background
2. Results
2.1. Properties of Mutational Signatures Observed in LUAD Patients
2.2. Pathway-Based Analysis and Relation between Signature Exposures and Gene Expression in Control Samples
2.2.1. Exposure to Smoking Signature Is Correlated with Increased Inflammatory Response in Non-Cancer Lung Tissue and Elevated Expression of the PD-L1 Immune Checkpoint Gene

2.2.2. Strength of SBS5, a Signature Correlated with Smoking but Not Unique to This Mutagen, Is Correlated with Changes in Ciliogenesis
2.2.3. Relation between the Strengths of APOEBEC-Related Signatures and Gene Expression
2.3. Mutational Signatures Reveal Relation between Exposure to Exogenous Processes and a Remodeling of Cell-Type Composition in Lung
3. Conclusions
4. Methods
4.1. Mutational Signatures
4.2. Expression Data
4.3. Clustering
4.4. Cell Composition Analysis with CIBERSORTx
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gorber, S.C.; Schofield-Hurwitz, S.; Hardt, J.; Levasseur, G.; Tremblay, M. The accuracy of self-reported smoking: A systematic review of the relationship between self-reported and cotinine-assessed smoking status. Nicotine Tob. Res. 2009, 11, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Tobacco Smoke Carcinogens and Lung Cancer. JNCI J. Natl. Cancer Inst. 1999, 91, 1194–1210. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; Sausville, E.L.; Girish, V.; Yuan, M.L.; Vasudevan, A.; John, K.M.; Sheltzer, J.M. Cigarette Smoke Exposure and Inflammatory Signaling Increase the Expression of the SARS-CoV-2 Receptor ACE2 in the Respiratory Tract. Dev. Cell 2020, 53, 514–529. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Taneja, V.; Vassallo, R. Cigarette Smoking and Inflammation. J. Dent. Res. 2011, 91, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Cortellini, A.; Giglio, A.D.; Cannita, K.; Cortinovis, D.L.; Cornelissen, R.; Baldessari, C.; Giusti, R.; D’Argento, E.; Grossi, F.; Santoni, M.; et al. Smoking status during first-line immunotherapy and chemotherapy in NSCLC patients: A case–control matched analysis from a large multicenter study. Thorac. Cancer 2021, 12, 880–889. [Google Scholar] [CrossRef]
- Li, J.J.; Karim, K.; Sung, M.; Le, L.W.; Lau, S.C.; Sacher, A.; Leighl, N.B. Tobacco exposure and immunotherapy response in PD-L1 positive lung cancer patients. Lung Cancer 2020, 150, 159–163. [Google Scholar] [CrossRef]
- Norum, J.; Nieder, C. Tobacco smoking and cessation and PD-L1 inhibitors in non-small cell lung cancer (NSCLC): A review of the literature. ESMO Open 2018, 3, e000406. [Google Scholar] [CrossRef]
- Sun, L.Y.; Cen, W.J.; Tang, W.T.; Long, Y.K.; Yang, X.H.; Ji, X.M.; Yang, J.J.; Zhang, R.J.; Wang, F.; Shao, J.Y.; et al. Smoking status combined with tumor mutational burden as a prognosis predictor for combination immune checkpoint inhibitor therapy in non-small cell lung cancer. Cancer Med. 2021, 10, 6610–6617. [Google Scholar] [CrossRef]
- Desrichard, A.; Kuo, F.; Chowell, D.; Lee, K.W.; Riaz, N.; Wong, R.J.; Chan, T.A.; Morris, L.G.T. Tobacco Smoking-Associated Alterations in the Immune Microenvironment of Squamous Cell Carcinomas. JNCI J. Natl. Cancer Inst. 2018, 110, 1386–1392. [Google Scholar] [CrossRef]
- Lafuente-Sanchis, A.; Zúñiga, Á.; Estors, M.; Martínez-Hernández, N.J.; Cremades, A.; Cuenca, M.; Galbis, J.M. Association of PD-1, PD-L1, and CTLA-4 Gene Expression and Clinicopathologic Characteristics in Patients With Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2017, 18, e109–e116. [Google Scholar] [CrossRef]
- Wang, G.Z.; Zhang, L.; Zhao, X.C.; Gao, S.H.; Qu, L.W.; Yu, H.; Fang, W.F.; Zhou, Y.C.; Liang, F.; Zhang, C.; et al. The Aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat. Commun. 2019, 10, 1125. [Google Scholar] [CrossRef] [PubMed]
- Basu, A. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef] [PubMed]
- Poon, S.; McPherson, J.R.; Tan, P.; Teh, B.; Rozen, S.G. Mutation signatures of carcinogen exposure: Genome-wide detection and new opportunities for cancer prevention. Genome Med. 2014, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Kim, Y.A.; Leiserson, M.D.; Moorjani, P.; Sharan, R.; Wojtowicz, D.; Przytycka, T.M. Mutational Signatures: From Methods to Mechanisms. Annu. Rev. Biomed. Data Sci. 2021, 4, 189–206. [Google Scholar] [CrossRef]
- Koh, G.; Degasperi, A.; Zou, X.; Momen, S.; Nik-Zainal, S. Mutational signatures: Emerging concepts, caveats and clinical applications. Nat. Rev. Cancer 2021, 21, 619–637. [Google Scholar] [CrossRef]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 3292. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.A.; Wojtowicz, D.; Sarto Basso, R.; Sason, I.; Robinson, W.; Hochbaum, D.S.; Leiserson, M.D.M.; Sharan, R.; Vadin, F.; Przytycka, T.M. Network-based approaches elucidate differences within APOBEC and clock-like signatures in breast cancer. Genome Med. 2020, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.T.; Synnott, N.C.; Rosenbaum, J.; Zhang, T.; Zhu, B.; Shi, J.; Zhao, W.; Kebede, M.; Sang, J.; Choi, J.; et al. Tracing Lung Cancer Risk Factors Through Mutational Signatures in Never-Smokers. Am. J. Epidemiol. 2020, 190, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2,658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Jones, P.H.; Wedge, D.C.; Sale, J.E.; Campbell, P.J.; Nik-Zainal, S.; Stratton, M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015, 47, 1402–1407. [Google Scholar] [CrossRef]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef]
- Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Health 2018, 15, 1033. [Google Scholar] [CrossRef]
- Kim, J.; Mouw, K.W.; Polak, P.; Braunstein, L.Z.; Kamburov, A.; Tiao, G.; Kwiatkowski, D.J.; Rosenberg, J.E.; Van Allen, E.M.; D’Andrea, A.D.; et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 2016, 48, 600–606. [Google Scholar] [CrossRef]
- Vieira, V.C.; Soares, M.A. The Role of Cytidine Deaminases on Innate Immune Responses against Human Viral Infections. BioMed Res. Int. 2013, 2013, 683095. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, D.S.; O’Donnell, D.; O’Connell, F.; O’Byrne, K.J. The role of inflammation in the pathogenesis of non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 2024–2036. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Liang, C.L.; Liu, H.; Zeng, Y.Q.; Hou, S.; Huang, S.; Lai, X.; Dai, Z. Impacts of cigarette smoking on immune responsiveness: Up and down or upside down? Oncotarget 2017, 8, 268–284. [Google Scholar] [CrossRef] [PubMed]
- Nomi, K.; Hayashi, R.; Ishikawa, Y.; Kobayashi, Y.; Katayama, T.; Quantock, A.J.; Nishida, K. Generation of functional conjunctival epithelium, including goblet cells, from human iPSCs. Cell Rep. 2021, 34, 108715. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.X.G.; Nakanaga, T.; Nadel, J.A. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-α-converting enzyme in human airway epithelial (NCI-H292) cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 287, L420–L427. [Google Scholar] [CrossRef] [PubMed]
- Bilsborough, J.; Viney, J.L. GPR15: A tale of two species. Nat. Immunol. 2015, 16, 137–139. [Google Scholar] [CrossRef]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef]
- Ai, L.; Chen, J.; Yan, H.; He, Q.; Luo, P.; Xu, Z.; Yang, X. Research Status and Outlook of PD-1/PD-L1 Inhibitors for Cancer Therapy. Drug Des. Dev. Ther. 2020, 14, 3625–3649. [Google Scholar] [CrossRef]
- Atchison, W.D. Effects of toxic environmental contaminants on voltage-gated calcium channel function: From past to present. J. Bioenerg. Biomembr. 2003, 35, 507–532. [Google Scholar] [CrossRef]
- Schuller, H.M. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar] [CrossRef]
- Goldfarbmuren, K.C.; Jackson, N.D.; Sajuthi, S.P.; Dyjack, N.; Li, K.S.; Rios, C.L.; Plender, E.G.; Montgomery, M.T.; Everman, J.L.; Bratcher, P.E.; et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat. Commun. 2020, 11, 2485. [Google Scholar] [CrossRef]
- Nemajerova, A.; Kramer, D.; Siller, S.S.; Herr, C.; Shomroni, O.; Pena, T.; Gallinas Suazo, C.; Glaser, K.; Wildung, M.; Steffen, H.; et al. TAp73 is a central transcriptional regulator of airway multiciliogenesis. Genes Dev. 2016, 30, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Licciardi, P.V.; Karagiannis, T.C. Regulation of Immune Responses by Histone Deacetylase Inhibitors. ISRN Hematol. 2012, 2012, 690901. [Google Scholar] [CrossRef]
- Schamberger, A.C.; Staab-Weijnitz, C.A.; Mise-Racek, N.; Eickelberg, O. Sci RepCigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci. Rep. 2015, 5, 8163. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Haswell, L.E.; Hewitt, K.; Thorne, D.; Richter, A.; Gaça, M.D. Cigarette smoke total particulate matter increases mucous secreting cell numbers in vitro: A potential model of goblet cell hyperplasia. Toxicol. In Vitro 2010, 24, 981–987. [Google Scholar] [CrossRef]
- Lumsden, A.B.; McLean, A.; Lamb, D. Goblet and Clara cells of human distal airways: Evidence for smoking induced changes in their numbers. Thorax 1984, 39, 844–849. [Google Scholar] [CrossRef]
- Damiá, A.d.e.D.; Gimeno, J.C.; Ferrer, M.J.; Fabregas, M.L.; Folch, P.A.; Paya, J.M. Arch BronconeumolA study of the effect of proinflammatory cytokines on the epithelial cells of smokers, with or without COPD. Arch. Bronconeumol. 2011, 47, 447–453. [Google Scholar] [CrossRef]
- de la Iglesia, J.V.; Slebos, R.J.; Martin-Gomez, L.; Wang, X.; Teer, J.K.; Tan, A.C.; Gerke, T.A.; Aden-Buie, G.; van Veen, T.; Masannat, J.; et al. Effects of Tobacco Smoking on the Tumor Immune Microenvironment in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2020, 26, 1474–1485. [Google Scholar] [CrossRef]
- Kim, V.; Oros, M.; Durra, H.; Kelsen, S.; Aksoy, M.; Cornwell, W.D.; Rogers, T.J.; Criner, G.J. Chronic bronchitis and current smoking are associated with more goblet cells in moderate to severe COPD and smokers without airflow obstruction. PLoS ONE 2015, 10, e0116108. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Lawrence, M.S.; Benes, C.H.; Zou, L. APOBEC3A and APOBEC3B Activities Render Cancer Cells Susceptible to ATR Inhibition. Cancer Res. 2017, 77, 4567–4578. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jia, M.; He, Z.; Liu, X.S. APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene 2018, 37, 3924–3936. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.W.; Gout, A.M.; Zhang, J. Therapeutic and prognostic insights from the analysis of cancer mutational signatures. Trends Genet. 2022, 38, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.L.; Li, Q.Y.; Tan, Q.Y. Smoking history and the efficacy of immune checkpoint inhibitors in patients with advanced non-small cell lung cancer: A systematic review and meta-analysis. J. Thorac. Dis. 2021, 13, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Boichard, A.; Tsigelny, I.F.; Kurzrock, R. High expression of PD-1 ligands is associated with Kataegis Mutat. Signat. APOBEC3 Alterations. OncoImmunology 2017, 6, e1284719. [Google Scholar] [CrossRef]
- Kim, V.; Jeong, S.; Zhao, H.; Kesimer, M.; Boucher, R.C.; Wells, J.M.; Christenson, S.A.; Han, M.K.; Dransfield, M.; Paine, R.; et al. Current smoking with or without chronic bronchitis is independently associated with goblet cell hyperplasia in healthy smokers and COPD subjects. Sci. Rep. 2020, 10, 20133. [Google Scholar] [CrossRef]
- Huang, X.; Wojtowicz, D.; Przytycka, T.M. Detecting presence of mutational signatures in cancer with confidence. Bioinformatics 2018, 34, 330–337. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Observation From Mutational Signatures | Supporting Literature |
|---|---|
| Cluster 2: | |
| ABOBEC signatures are associated with expression of SFTPB and SFTPC | novel observation |
| APOBEC might indirectly trigger the expression PD-1 | [56] |
| Cluster 5: | |
| Smoking triggers pro-inflammatory response and cytokines signaling | [28,32] |
| Smoking increases MUC5AC expression | [34] |
| Smoking increases PD-L1 expression | [36] |
| Smoking increases GPR15 expression | [35] |
| Cluster 6: | |
| APOBEC is associated with a reduction in cilium organization | novel observation |
| Cell-type composition: | |
| ABOBEC signatures are associated with a reduction in CD8+ cells | [48] |
| Smoking is associated with increase of goblet cells | [45,46,57], |
| Smoking is associated with decrease of ciliated cells | [3,40,41] |
| APOBEC is associated with decrease of ciliated cells | novel observation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.-A.; Hodzic, E.; Amgalan, B.; Saslafsky, A.; Wojtowicz, D.; Przytycka, T.M. Mutational Signatures as Sensors of Environmental Exposures: Analysis of Smoking-Induced Lung Tissue Remodeling. Biomolecules 2022, 12, 1384. https://doi.org/10.3390/biom12101384
Kim Y-A, Hodzic E, Amgalan B, Saslafsky A, Wojtowicz D, Przytycka TM. Mutational Signatures as Sensors of Environmental Exposures: Analysis of Smoking-Induced Lung Tissue Remodeling. Biomolecules. 2022; 12(10):1384. https://doi.org/10.3390/biom12101384
Chicago/Turabian StyleKim, Yoo-Ah, Ermin Hodzic, Bayarbaatar Amgalan, Ariella Saslafsky, Damian Wojtowicz, and Teresa M. Przytycka. 2022. "Mutational Signatures as Sensors of Environmental Exposures: Analysis of Smoking-Induced Lung Tissue Remodeling" Biomolecules 12, no. 10: 1384. https://doi.org/10.3390/biom12101384
APA StyleKim, Y. -A., Hodzic, E., Amgalan, B., Saslafsky, A., Wojtowicz, D., & Przytycka, T. M. (2022). Mutational Signatures as Sensors of Environmental Exposures: Analysis of Smoking-Induced Lung Tissue Remodeling. Biomolecules, 12(10), 1384. https://doi.org/10.3390/biom12101384