1. Introduction
In 1988, in a first for genetically linked disorders, Wallace et al. [
1] described the potential link between a rare optic neuropathy and a mutation in mitochondrial DNA (mtDNA). The disease, known as Leber hereditary optic neuropathy (LHON), eponymously named after the physician who first described it in 1873 [
2], is a neurodegenerative disorder that causes bilateral blindness by causing the death of retinal ganglion cells (RGCs). Although it can occur at any age, it primarily affects young adults [
3]. LHON is a clinical diagnosis that begins with the onset of a cecocentral scotoma, though it is now definitively assessed by DNA testing for known mtDNA mutations [
4]. Once started, vision loss happens rapidly over weeks or months, and although it usually begins in one eye, the contralateral eye inevitably follows [
4]. All LHON cases have predominately homoplastic mitochondrial DNA point mutations that affect complex I subunits [
1,
5,
6]. Three mutations are most prevalent (m.11778G > A in ND4, m.14484T > G in ND6, and m.3460G > A in ND1) and account for 95% of cases [
7]. No known nuclear DNA mutations cause LHON [
8], yet interplay with nuclear genetic factors is likely [
9]. The deleterious effects of mtDNA mutations have also been established in animal models through transferring mutant human mtDNA ND4 [
10] or mutant mtDNA ND6 genes [
11] to mice, which caused visual loss similar to the effects experienced by LHON patients. Although the genetic defects are a necessary precondition for the disease, as the mitochondrial mutations are present in all LHON-affected patients, not all carriers of the defects exhibit the illness [
12]. Penetrance of the disease is low in female carriers of the mutations [
13], and a mitochondrial biogenesis compensatory mechanism is present in both carriers and affected cases [
12]. People that have any of the LHON mitochondrial mutations exhibit more copies of their mtDNA than control cases [
12]. Affected individuals, although they show more mtDNA per cell than controls, exhibit less mtDNA than carriers, implying a reduced mitochondrial compensation capacity [
12].
Much progress has been made in unravelling LHON pathophysiology, molecular and environmental triggers, but the causal chain leading to the illness and the roots of its sudden onset and progression are still unelucidated [
4]. While neuroretinal effects of the disease are well established [
14,
15], and studies of postmortem optic nerve samples have been published [
16], the actual dynamics of injury propagation in RGCs and the biochemistry of the process that leads to RGC apoptosis are still unknown [
7,
17].
LHON-linked mtDNA mutations affect critical components of the mitochondrial electron transport chain, and, as a result, energetic imbalance and oxidative stress have been used to explain the death of affected RGCs [
18,
19]. The clinical findings in LHON (loss of visual acuity, cecocentral visual field loss, and dyschromatopsia) reflect the loss of small diameter axons, which led the Sadun group [
16,
17,
20] to hypothesize that the axonal diameter predisposes the axon to death because of energy imbalance. In this hypothesis, and consistent with the axonal stress index (NFL-SI) that the Sadun group developed [
16], axons undergo degeneration because of an imbalance between energy requirements, which are proportional to the axonal surface, and ATP production, which is proportionate to axonal volume. The axonal stress index (NFL-SI) is a good predictor of the bias towards injury of smaller diameter axons in LHON. However, the disease also has axonal loss topological characteristics that cannot be explained by the index alone. In particular, the ATP deficit hypothesis and the NFL-SI index can account for increased death susceptibility of smaller axons but does not account for the clustering of RGC loss seen in LHON optic nerve histology [
21].
Experimental data from rats demonstrates that, in optic nerve axons, mitochondrial volume is directly proportional to the axon section volume in both myelinated and unmyelinated segments [
22]. However, in cybrids with LHON mutations, overall cellular ATP production is normal, although ATP production per mitochondrion is impaired [
23,
24]. “However, in cybrids with LHON mutations, overall cellular ATP production is normal, although ATP production per mitochondrion is impaired [
23,
24]. Animal models with the 14484T > G (ND6) mutation, created by the Sadun group [
24], and models with the 3460G > A (ND1) mutation, created by Zhang et al. [
23], maintain levels of ATP production but cellular ROS levels increase. Maintaining overall cell ATP production can be explained by a mitochondrial compensatory mechanism [
12], where the mitochondrial volume increases, compensating for the lower output per mitochondrion.
The role of reactive oxygen species (ROS) in the genesis and evolution of LHON is a promising alternative to the ATP deficit hypothesis [
8,
25]. Mitochondria are the site of oxidative phosphorylation and the source of most cellular superoxide (
production [
26,
27], a precursor to all ROS compounds [
28]. Although ROS compounds have intracellular signaling roles in fundamental cellular processes [
29,
30], it has also been recognized that higher concentrations lead to oxidative stress and can trigger cell death [
31]. We used both
and hydrogen peroxide (H
2O
2) as a stress-inducing species, recognizing that H
2O
2 physical properties make it a more likely candidate as the oxidative stress species. The range of ROS concentrations with beneficial physiological outcomes is relatively narrow. For example, H
2O
2 concentrations ([H
2O
2]) between 1 nM to 10 nM benefit neuronal development, while concentrations below 1 nM and over 100 nM led to tissue pathology [
32].
There is experimental evidence that increases in ROS concentration are involved in LHON pathophysiology, although the actual mechanism mediating this process is unknown. For example, in animal models, ROS production increases by 48% when the 14484T > G (ND6) mutation is present [
24], and by about 30% when the 3460G > A (ND1) mutation is present [
23]. Data also points to lower antioxidant defenses in LHON cases, as exhibited by cybrids carrying LHON mutations, which can also lead to increased ROS [
19]. It has also been shown that cigarette toxicity, often considered an environmental factor in the emergence of LHON, reduces ROS scavenging capacity [
13]. Searches for therapeutic measures to prevent or stop its development have also concentrated on antioxidants and oxidative stress-reducing medication, although other approaches targeting estrogen receptors [
33] and gene therapy have recently been studied [
34]. Currently, the only marketed LHON therapy (authorized under exceptional circumstances in Europe) is idebenone, a synthetic analogue of CoQ10 that reduces oxidative stress [
34], although the mechanism by which this action is achieved is unclear [
35,
36].
The basis for the present study is the hypothesis that LHON axonal degeneration has specific characteristics that might be explained by ROS spreading from one axon to another. Analysis of axonal diameter loss shows that, although smaller diameters are affected disproportionately, the effect highly depends on the optic nerve areas where the loss occurs [
16]. Small optic nerve fibres are alive in unaffected areas, while larger diameter RGCs are dead in affected areas, which show complete RGC loss [
16]. In other words, the loss of axons is area-specific and not solely diameter-specific.
We previously modeled the wave-like propagation of LHON [
37], with a good correlation between clinically observed patterns of RGC loss and the size-dependent model presented by the Sadun group [
16]. Our previous model [
37] operated on a 2-dimensional (2D) section of the optic nerve and studied the propagation of axonal degeneration in the optic nerve from a selected initial point of injury. The present study extends the modeling from [
37] 2D to 3 dimensions (3D), adds axonal membrane properties, a quantitative biochemical model, a more complex biological model for axonal sections, and the ability to generate ROS outside the axonal space. The 3D model also allows the study of processes around the nodes of Ranvier, present in the myelinated part of the optic nerve [
38], calculates the time spent in the sample’s biology, allowing us to judge the timescales of the processes under study. With these enhancements, we studied the connection between the mitochondrial volume increases that are present in carriers and afflicted LHON patients [
12]. We also studied under what conditions axonal fibers degenerate independently and when degeneration is zonal. Finally, we investigated possible triggers for the wave-like propagation of the illness. Both
and H
2O
2 were used as oxidative agents. Finally, an unresolved question is the location of the initial injury for LHON along the optic nerve. The present study explored if the injury starts in the unmyelinated prelaminar region or the myelinated region of the optic nerve, using a computational approach.
3. Results
3.1. Superoxide Alone Cannot Explain LHON Injury Patterns
Superoxide is the primary ROS generated by complex I, and its production is increased in LHON cybrids [
23,
24]. However,
is an electrically charged species that does not diffuse through cellular membranes [
61] and can be dismutated to H
2O
2. To test if
can explain the zonal loss patterns observed in LHON pathologies, we ran simulations that generated
only intra-axonally and did not allow
to migrate to the inter-axonal space (i.e., Pm set to 0 µm s
−1). We set the
concentration threshold at which axonal degeneration would occur to 10
−1 nM because reported cellular concentrations of
are in the 10
−2 nM range [
47], and there are no known concentration thresholds that would trigger pathological issues because
is rapidly converted to H
2O
2 in vivo [
42]. Given the lack of knowledge about whether stressed axons produced more or less ROS than health axons, we compared the results of simulations where there was increased (PF
RP) or decreased (NF
RP)
production, to check the sensitivity of results to ROS production changes in the Stress state.
Loss patterns were similar irrespective of the mitochondrial ROS production patterns because axonal loss depends only on each axon’s ROS production and is not influenced by superoxide diffusion from other cells (
Figure 4a,b). However, if all mitochondria produced ROS at the same rate (RP
SAME), larger diameter axons were affected in higher numbers (
Figure 4a simulations marked with RP
SAME), which is uncharacteristic for LHON. The axonal loss was uniform across all optic nerve octants (
Figure 4b), and at the end of simulations, each octant had more mid-diameter axons alive than small or large diameters. The loss patterns were also atypical for LHON [
16,
37]. Interestingly, although all simulations presented in
Figure 4a had similar surviving axonal ratios (around −6dB for each octant), they had markedly different patterns in the coronal sections of the optic nerve, with the experiments where larger diameter axons degenerated showing a more sparse appearance (i.e., a lot more “blue” areas in
Figure 4a) Although less numerous, larger diameter axons occupied significant portions of the overall optic nerve area. If they are affected, a significant portion of the optic nerve area loses neurons, and the remaining axons are sparsely distributed among areas where no other axons are present. When mitochondria produced
at the same rates, larger diameter axons were disproportionately affected (
Figure 4c,d).
When
production uses the constant neuronal firing frequency hypothesis (RPF
CONST), axonal loss is heavily correlated with the axonal diameter and skewed towards lower diameter axons. The affected axons demonstrated extreme diameter selectivity (
Figure 4c), with smaller axons completely degenerating while larger diameters were unaffected. Thus, optic nerve octants were affected in proportion to their smaller diameter axons. Surviving and dead axons intersperse, with no area of complete axonal loss. The observed patterns differed from actual LHON axonal loss patterns, which show a total loss in large areas, regardless of axonal diameters [
16,
37].
Thus, given simulation results that could not replicate actual LHON injury patterns because of the inability of to diffuse across cell membranes, it is unlikely that is the primary axonal damage agent in LHON. Because of the permeability of H2O2 through cell membranes, subsequent simulations focused on it as a potential oxidative stress mediator for axonal degeneration in LHON.
3.2. The Constant Neuronal Firing Frequency Hypothesis Predicts Higher Oxidative Stress in the Temporal Optic Nerve
We considered two ROS production hypotheses, one in which all mitochondria produced ROS at the same rate (RP
SAME), and one where mitochondria produced ROS in proportion to the axonal energy requirements (RPF
CONST). To determine which of the two production models was more relevant to LHON, we ran simulations with identical initial topology and biochemistry but with different H
2O
2 production models and measured the oxidative stress produced by the mitochondria. Both unmyelinated (
Figure 5a) and myelinated (
Figure 5b) topological models were used for this purpose. Axonal mitochondria volume ratio (mito%) was measured as a proportion of the axonal volume it occupies [
22]. Simulations were calibrated at 4% mito% in the unmyelinated region [
22] so that, regardless of the ROS production strategy, mean H
2O
2 concentrations were the same (
Figure 5a).
All simulations demonstrated the formation of nonuniform H
2O
2 concentration gradients in the nerve, ranging from 5–100 nM. Axonal mitochondrial volume ratios (mito%) influenced the gradients in ways that depended on the production model and the myelination of the region (
Figure 5). When all mitochondria produced H
2O
2 at the same rate (RP
SAME), H
2O
2 concentrations increased as mito% increased (
Figure 5 and
Figure 6). This phenomenon happened in both unmyelinated and myelinated regions because the total H
2O
2 produced was proportional to the total amount of mitochondria present when the rate of mitochondrial ROS production was fixed. Under this production model, carriers of LHON mutations would experience higher oxidative stress than LHON cases. Given that localized H
2O
2 concentrations can be higher than the mean concentration, many axons could experience high oxidative stress levels, increasing the probability of axonal degeneration (
Figure 6c). The constant neuronal firing frequency assumption (RPFCONST) demonstrated inverse correlations between mean H
2O
2 concentrations and mito%.
Other differences between the RPFCONST and RPSAME production models were observed. Higher concentrations of
in the temporal nerve relative to the nasal nerve were observed in all simulations that used the RPFCONST hypothesis (
Figure 6), while in the RPSAME production hypothesis, these zonal gradients were undetectable for mito% above 4% (
Figure 6c). The axonal diameter was positively correlated with oxidative stress in the RPSAME cases (
Figure 6c) and inversely correlated in the RPFCONST cases (
Figure 6c).
Increasing the axonal membrane permeability allowed higher rates of ROS diffusion between the axon and the extra-axonal space, which reduced [H2O2] gradients in unmyelinated and myelinated model simulations because it moved the H2O2 from its mitochondrial source in the axon to a larger scavenging volume. The differences between the mean ROS concentration and the distribution of individual voxel concentrations illustrate the challenge of using means as a figure of merit in this case. For example, H2O2 concentrations inside axons can be above the threshold for degeneration, yet the mean concentration across the entire simulated volume can be significantly below those values. Thus locally, H2O2 can damage axons, while the axons remain intact in other parts of the model.
Under the hypothesis that high H
2O
2 concentrations trigger axonal degeneration, the constant mitochondria production assumption (RP
SAME) creates optic nerve ROS levels that would result in more extensive optic nerve damage when the mito% is increased. However, LHON carriers have higher mito% than LHON cases, even though both have the same mutation [
12], implying a lower oxidative load on the optic nerve. Thus, the RP
SAME production model is unlikely because it does not fit the data from studying LHON axonal mitochondria volume ratios (mito%) and was not studied in subsequent simulations.
3.3. Unmyelinated Regions of the Optic Nerve Have Higher ROS Levels Than Myelinated Regions
The optic nerve has a narrow geometry, with a diameter less than 3% of its length. In geometries where one dimension is significantly larger than others, diffusion processes proceed faster on the smaller dimensions. Therefore, lengthy areas where scavenging is present without significant ROS production might reduce the influence that high oxidative stress regions can have on their surroundings. We hypothesized that H2O2 concentration values and their changes in time in the unmyelinated region are largely decoupled from the [H2O2] in the myelinated area.
To study the hypothesis, simulations were performed with an abridged optic nerve (X = 150 µm, Y = 150 µm, and Z = 410 µm) containing myelinated and unmyelinated regions. Along the Z axis, the top 50 µm modeled the unmyelinated region, followed by 200 µm axonal myelinated region, a 5 µm section comprising a node and another 155 µm region modeling a myelinated axonal shaft. The node’s length was larger than the published data from rats [
53] because of limitations in the simulation setup for samples of this scale. The internodal sizes were between reported values [
53]. Nodes from all axons were aligned in the same plane, which is not physiological, but serves to increase oxidative coupling between axons, thus creating a more oxidative environment than in vivo. Therefore, this limitation should not have changed the qualitative nature of the results.
In this simulation, the role of scavenging rates along the axon was also examined. For this purpose, either similar scavenging rates across the model (
Figure 7) or a 30% of unmyelinated scavenging capacity in the myelinated regions (
Figure 8) were assumed.
Simulations were run until [H
2O
2] reached a steady state. The unmyelinated regions had significantly higher [H
2O
2] than the myelinated regions for both uniform scavenging of H
2O
2 (
Figure 7) and reduced H
2O
2 scavenging in the myelinated area (
Figure 8). In both cases, coronal plane views (
Figure 7a and
Figure 8a) demonstrate [H
2O
2] gradients between the temporal and nasal sides, with higher [H
2O
2] on the temporal side of the nerve.
Transverse (
Figure 7b and
Figure 8b) and sagittal (
Figure 7c and
Figure 8c) views demonstrate significantly higher gradients along the Z axis than in the XY plane. H
2O
2 concentrations in the myelinated and the unmyelinated regions were higher than the internodal concentrations when scavenging enzymes were uniformly distributed in the sample (
Figure 7b,c).
The mitochondria in the internodal region had lower H
2O
2 concentrations than the paranodal mitochondria, which support signal conduction by providing ATP to the node (
Figure 7 and
Figure 8). The decrease in internodal scavenging increased H
2O
2 concentrations in the internodal regions, while concentrations in other regions were similar and higher than in the internodal regions (
Figure 8). Coronal views of the unmyelinated region show no change in H
2O
2 concentrations (top row of
Figure 7a and
Figure 8a), although a coronal section of the node shows a small increase (third row of
Figure 7a and
Figure 8a).
In both cases, H2O2 concentrations in the internodal regions are lower than the values reached in the unmyelinated region or at the node. As a result, the H2O2 gradients are towards the internodal regions, and no gradient connects the myelinated and unmyelinated regions. Therefore, decomposing the unmyelinated and myelinated regions into separate models would not affect results.
The results from these simulations enable the separation of a nerve into separate models of its myelinated and unmyelinated regions, running the simulations on the 2 models using the same biochemical setup and analyzing the results as if they were from a unified model. Such a separation is useful for simulation throughout because large models have high computational requirements.
3.4. Myelinated Regions Are Unlikely Locations for Initiating Axonal Degeneration in LHON
The region where LHON optic nerve lesions are initiated and the dynamics of axonal degeneration associated with the disease are unknown. Under the hypothesis that the disease is triggered by oxidative stress in the axonal region of the optic nerve, we asked if the myelinated or the unmyelinated regions are more prone to high oxidative stress. Intuitively, higher axonal mitochondrial volume ratios (mito%) in the unmyelinated region [
22,
52] should have higher concentrations of ROS. However, as shown previously, the ROS gradients within the confines of the axon can be very high and vary based on the axon diameter. We also showed that when mitochondria produce ROS in proportion with the axonal energy requirements, overall H
2O
2 levels decrease as the axonal mito% increases. The myelinated region membranes are also less permeable to H
2O
2 because of the many myelin layers surrounding them. From that perspective, the oxidative environment in the myelinated regions is greater. However, previous simulation results in the myelinated and unmyelinated models showed higher oxidative stress in the unmyelinated region. Thus, we hypothesized that mitochondria in the unmyelinated regions are more likely to suffer from higher levels of oxidative stress, potentially triggering axonal degeneration and subsequent neuronal apoptosis.
We tested this hypothesis on unmyelinated and myelinated models, running simulations that considered only the effects of H
2O
2 produced by axons while varying the mitochondrial H
2O
2 in the Stress state (
Figure 9 and
Figure 10). Myelinated region models were created as coronal slices around a node, with an overall model thickness of 10 µm. A single node was placed 6 µm from the proximal side of the model. Mitochondria were placed symmetrically above and below the node, with no mitochondria present at the node.
Signal conduction energy requirements represent approximately 75% of neuronal energy requirements, with the rest used for homeostatic cellular functions [
67,
68]. The myelinated segment of the optic nerve dominates the axon length, with internodal distances between 100–200 µm for each node of 1–2 µm [
53]. Therefore, in axonal segments centered on a node, most internodal mitochondria produce 25% of the required ATP, while the mitochondria close to the node produce 75% of the energetic needs of the segment. Assigning an estimated 5% ROS production rate to the internodal mitochondria farther from the node should account for the energy requirements of that segment.
To further explore myelinated vs. unmyelinated axons, we again assessed two models for oxidatively stressed axons, one with increased oxidative production in the Stress state (PF
RP) and one with decreased production (NF
RP). Simulations were performed to test the possibility that mitochondria change their operating points when subject to oxidative stress. For the PF
RP cases, we ran simulations where the Stress and Healthy states had either the same production values (
Figure 10a) or increased production values (
Figure 10b). To test sensitivity to membrane permeability, we used two different H
2O
2 membrane permeability values (P
m = 20 µm s
−1 and P
m = 200 µm s
−1).
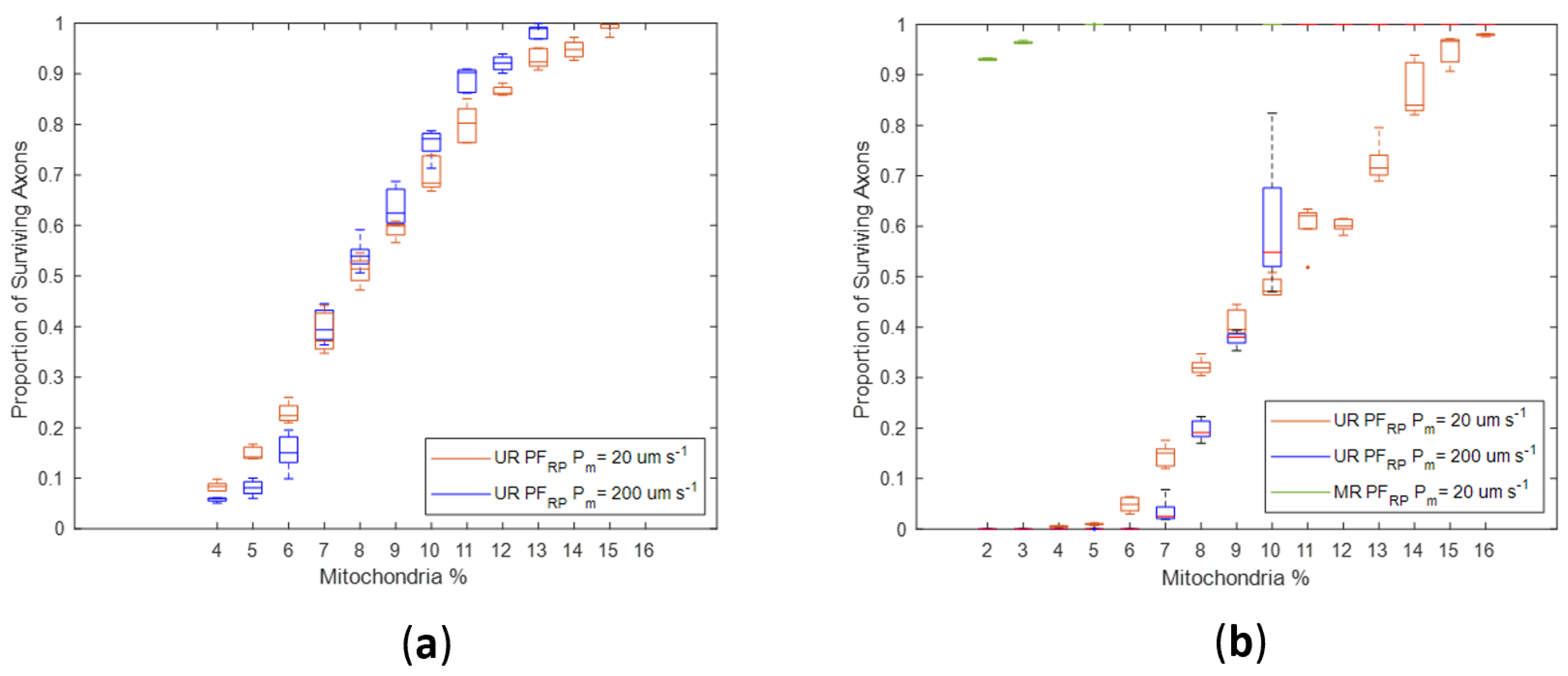
Regardless of H
2O
2 production changes in the Stress state, the proportion of axonal loss was significantly lower in simulations of myelinated axons than in unmyelinated axons (
Figure 9 and
Figure 10). Notably, there was no axonal loss in the myelinated regions in NF
RP simulations (
Figure 9) or PF
RP simulations (
Figure 10a). In PF
RP simulations with increased stress levels, myelinated regions experienced loss only for the lower membrane permeability value.
In unmyelinated regions, the higher energy needs imposed by less efficient axonal conduction led to higher H
2O
2 production rates. For unmyelinated models, even if H
2O
2 production was reduced in the Stress state (NF
RP), axonal degeneration occurred across a wide range of mito% values (4% to 15%) (
Figure 9). However, axonal survival increased linearly with mito% (
Figure 9).
Simulations where production in the Stress state stayed the same or increased (PF
RP) had greater variability in axonal survival than the NF
RP simulations for the same mito% (
Figure 9 and
Figure 10). For the PF
RP simulations, the proportion of surviving axons graphed against the mito% had a more sigmoid shape. The proportion of surviving axons changes more dramatically for small changes in mito% as the production of H
2O
2 increases in the Stress state (
Figure 10a,b). At the higher membrane permeability value (P
m = 200 µm s
−1) and for the case when production in the Stress state increases, there were abrupt changes in survival for small variations in mito% around the 10% mito% value (
Figure 10b). In this case, slight topological changes resulting from re-running the axonal placement algorithm for each run result in greatly different axonal survival ratios. The sensitivity of results to small axonal placement changes suggests that the interplay between axonal topology, membrane permeability and mito% are relevant to oxidative injury to axons.
Membrane permeability increases oxidative linkage between adjacent axons. Surprisingly, higher membrane permeability increases axonal losses at lower mito% and decreases axonal loss at higher mito% (
Figure 9 and
Figure 10). This was true for both Stress state production models. Specifically, in the NF
RP production model, a higher membrane permeability decreases axonal loss when mito% > 5% (
Figure 9), in the Stress = Health model when mito > 7% (
Figure 10a), and in the PF
RP model when mito% > 10% (
Figure 10b). The existence of a mito% value below which high membrane permeability is a deleterious factor in axonal survival and above which membrane permeability has a positive effect demonstrates that the many factors which contribute to the subcellular oxidative environment interact with each other in nonobvious ways. In this case, the interaction between membrane diffusion, optic nerve axonal topology, mito% and Stress state oxidative behavior nonlinearly affect simulation outcomes.
The simulation results suggest that myelinated portions of RGC axons are unlikely to be the starting point of the injury in LHON. It is more likely that the injury occurs in the unmyelinated portion of the optic nerve and that axonal membrane permeability to H2O2 creates an oxidative environment which leads to axonal loss. It is also probable that during the acute phase of the injury, mitochondria will produce the same or greater amounts of H2O2, thus intensifying the overall oxidative stress environment of the nerve.
3.5. Increasing Physiological Coupling between Axons Increases Localized Axonal Degeneration
Results from previous sections show that axonal degeneration was strongly correlated with axon diameter when the axonal membrane was impermeable to ROS (as with
). Each coronal optic nerve octant had surviving axons (
Figure 4b). However, if a ROS had sufficient membrane permeability to diffuse through the axonal membrane (as with H
2O
2), different axonal degeneration patterns occurred depending on ROS production in the Stress state (PF
RP vs. NF
RP;
Figure 11a,b).
The results also show that the interplay between several factors can create qualitatively different loss outcomes. Changes in ROS production or scavenging can change the oxidative state of the optic nerve and trigger oxidative degeneration in large areas of the optic nerve. Regardless of the Stress state oxidative behavior, the temporal side octants suffered heavier axonal losses (
Figure 11b). Nevertheless, the extent of the loss and the patterns depended heavily on the oxidative behavior in the Stress state.
If the intra-axonal H2O2 concentration was higher than outside the axon, permeability through the axonal membrane allowed the transport of H2O2 to the inter-axonal space. Conversely, if the intra-axonal H2O2 concentration was lower than outside the axon, axons were affected by extra-axonal H2O2. This oxidative connection between a neuron and its environment can be beneficial or deleterious. Under such conditions, adjacent axons create a localized oxidative environment. This oxidative interdependence can lead to axonal losses that are geometrically close, regardless of the diameters of the adjacent axons. Such topological loss patterns were called “neighborhood losses.”
Loss of neighboring axons was present regardless of the Stress state production whenever the membrane was permeable (
Figure 11c). For NF
RP simulations, the effect was less prevalent and not as visually striking due to its prevalence in areas where small and medium diameter axons are grouped and lack of injury beyond small and medium diameter axons. (NF
RP simulations in
Figure 11a). Areas of degeneration were many yet small (PF
RP marked simulations in
Figure 11a). This pattern is atypical for LHON pathology [
37].
In contrast, in simulations where H
2O
2 production increased in stressed axons (PF
RP), there was high neighborhood loss, and in most cases, the loss was complete in each octant (PF
RP simulations in
Figure 11). This injury pattern (
Figure 11a) resembles that seen in LHON pathology [
37]. Higher mito% provided more protection from injury and reduced the extent of the injury patterns in proportion to the axonal mitochondrial volume ratios (mito%). Giordano et al. [
12] found that carriers of the LHON mutation who do not suffer from visual loss have increased mito% relative to LHON patients. Although there is no data correlating injury extent with mito% present in axons, our model correctly predicts that there is a mito% where the injury does not appear. In all cases, axonal degeneration started in the temporal size of the optic nerve, which also correlates with initial visual loss in LHON [
16,
37] (
Figure 11a).
At the end of a simulation, when a steady state was reached after completion of axonal loss, the map of ROS concentration was lowest where the axonal loss was greatest, i.e., axonal loss contributed to lowering the oxidative stress in the tissue, in both the NF
RP and PF
RP paradigms (
Figure 11e).
In summary, the neighborhood loss effect was stronger in the PFRP paradigm, and this pattern better corresponded to LHON pathology, suggesting that no change or an increase in ROS production in stressed axons is more likely in the acute phase of LHON.
3.6. Glia-Produced Hydrogen Peroxide Produces Cascading Axonal Failure
Optic nerve glia are also possible sources of oxidative stress. Our previous study focused only on axonal-generated ROS [
37]. Given that axonal and glial mitochondria are assumed to have Stress states, there are 4 possible combinations: axon and glial mitochondria both use NF
RP in the Stress state, both use PF
RP, or they use opposite ROS production strategies. When both axonal and glial mitochondria used the same Stress state production model, the simulation results were qualitatively similar to the ones presented in the previous sections (
Figure 9,
Figure 10 and
Figure 11). Such results are expected because adding glial elements between axons is equivalent to adding axons with a virtual membrane fully permeable to H
2O
2. If axonal mitochondria used PF
RP and glial mitochondria used NF
RP, the results were also qualitatively close to the previous PF
RP studies (
Figure 10 and
Figure 11). Glia-produced H
2O
2 increases oxidative stress in all axons, exacerbating the loss experienced in PF
RP relative to the same simulations that do not consider glial elements.
The combination where axonal mitochondria produced less ROS in the Stress state (NF
RP mode) and glial mitochondria produced more ROS (PF
RP mode) was surprising (
Figure 12 and
Figure 13). Given that NF
RP simulations with no glia resulted in a weak neighborhood effect, we hypothesized that the addition of glia operating in PF
RP mode to unmyelinated axon models would increase the area of neighborhood loss, relative to the case where only axonal mitochondria were present. Indeed, we found that neighborhood effects extended over larger areas relative to the base case (i.e., NF
RP simulations without glia present), although the scale at which local loss occurs was significantly smaller than in the PF
RP cases (
Figure 12a,b). If glial mitochondrial content was increased, the neighborhood effect was stronger, with local injury extending to larger groups of adjacent axons (
Figure 12a). In all simulations where glial mitochondria were present, axonal loss was more prevalent in the temporal side of the model (
Figure 12), as was seen when glial mitochondria were absent.
Higher glial mitochondria content will increase the overall model H
2O
2 concentrations. The H
2O
2 concentrations maps (
Figure 12e) show the increase created by the glia-generated H
2O
2, generating a more uniform H
2O
2 concentration distribution across the model. Surprisingly, even under such homogeneous H
2O
2 concentrations, the temporal side was still more susceptible to oxidative damage, probably because of its higher density of small diameter axons. Although glia’s contribution to [H
2O
2] creates a more homogeneous oxidative environment, the per axon differences are still a factor (
Figure 12e).
Membrane permeability had a limited effect on the effects of glial ROS production models (
Figure 13), similar to cases where glial mitochondria were absent. However, the results with a 10% glia setting and lower axonal mito% proportions (4% and 5%) were independent of the membrane permeability (
Figure 13). When glia mito% were decreased, significantly greater percentages of axons survived (
Figure 12).
Because of membrane permeability in unmyelinated areas, H
2O
2 produced outside the axon will influence the axons and vice versa. Glia are present in the myelinated region but not in the areas next to the nodes [
12]. Myelinated axons are impermeable to H
2O
2 outside the node; therefore, ROS outside the axon would not penetrate the axon. Higher H
2O
2 levels could affect myelination, although our models do not capture such effects. Therefore, we only simulated unmyelinated optic nerve sections in the studies of axonal-glial interactions.
Overall, H2O2 created by glia can affect axons in unmyelinated regions, but is not necessary to explain the neighborhood loss patterns exhibited by LHON patients. Glia may have a supporting role in axonal degeneration, but the main oxidative engine is likely intra-axonal mitochondria.
4. Discussion
The mechanism for propagation of axonal degeneration in LHON is unknown, as is the location of the initial injury within the optic nerve. We previously described a 2-dimensional coronal plane propagation algorithm that produced injury patterns which correlated with clinically observed patterns of RGC loss [
37]. In the current study we expanded that work by adding 3-dimensional optic nerve section modelling, more realistic biophysical processes and parameters, use of membrane permeability, modeling of unmyelinated and myelinated axons, the addition of glia-produced ROS, and interpretation of biological vs. simulation time with respect to biology. The current work also allows the study of ROS generation and diffusion processes around the nodes of Ranvier, which are areas of more intense energy demands within the myelinated regions of the optic nerve.
We showed that H
2O
2 is more likely than
to be the primary ROS in propagating LHON injuries, owing to its cellular membrane impermeability and its fast dismutation to H
2O
2. This result has therapeutic implications. Increasing peroxidase activity in the optic nerve extra-axonal space could presumably decrease the propagation of the axonal degeneration and may be more amenable to drug delivery than delivering a SOD mimetic [
69,
70], which would have to enter the axon.
The study modeled and explored several hypotheses regarding the influence of H2O2 in LHON injury propagation dynamics. We found that when the axonal membrane is permeable to H2O2 and oxidative stress induces larger production of H2O2, small injuries extend to neighboring axons. Our work suggests that a limited systemic insult can affect a region of the optic nerve in a process akin to a selective axotomy, triggering axonal degeneration in the respective RGCs. When some axons die, a new equilibrium point is found. This can explain why LHON rarely results in complete blindness, instead primarily producing loss of central vision and clinical findings of small axon-diameter disease.
By studying the propagation of H2O2 in models that contained unmyelinated and myelinated regions, we showed that due to their relative distance, the two regions do not influence each other oxidatively. Specifically, the origin and propagation of axonal degeneration in this simulation model is biased toward greater loss in the unmyelinated portion of the nerve rather than the myelinated nerve. In other words, it is possible that there may be early oxidative injury to axons within the optic nerve head that is of a greater degree than the retrobulbar nerve. Given that the optic nerve head is clinically visible, then if swelling or other changes could be detected before visual loss occurs, there may be a therapeutic window before the rest of the nerve is injured and axons are lost. Oxidative axon injury restricted to the unmyelinated nerve but without propagation to the rest of the nerve could cause localized loss of axonal conduction and visual loss. If conduction is restored as a result of lower ROS levels, then this could represent a mechanism by which LHON spontaneously improves in many patients.
It is known that positive feedback (i.e., the ability of an external signal to increase its production) in different systems, e.g., ROS and calcium, can result in wavelike propagation [
54,
71]. However, all such wave-generating models have an external wave initiation event. For example, our previous model [
37] had to be triggered by an “injury stimulus.”. In contrast, the current study describes self-triggered injuries that appear because of an individual cell’s biochemistry in the context of its milieu. This may be relevant to patients with LHON who begin having visual loss soon after heavy drinking, smoking, or other stressors. LHON patients who chronically smoke are relatively protected and lose vision more insidiously [
14]. It is possible that ROS levels in smokers lead to early chronic loss of small diameter axons, similar to the NF
RP patterns of
Figure 11a. This effect might provide later protection by reducing the overall density of small axons in the optic nerve. The model would also allow simulating the effects of myelin development, mediators of systemic oxidative stress, and other parameters that could be assessed biophysically.
We found that H
2O
2 concentrations correlated with axonal degeneration patterns seen in LHON histological sections [
37]. Higher oxidative levels were present in the temporal side of the nerve before the onset of axonal loss, which was also on the temporal side. These findings correlate well with clinical observations [
2]. After axons have been affected, a new equilibrium is found where less H
2O
2 is produced. The degeneration of some axons and the subsequent cessation of ROS production can have a protective effect on the remaining axons. Clinical observations on children under 12 show that despite structural evidence of axonal loss, there is less functional loss and no loss later in life [
72]. The proportion of axonal volume occupied by (mitochondria volume ratio; mito%) heavily influenced the extent of degeneration, in line with clinical observations [
12]. Given that ROS have roles as signaling agents in the control of mitochondrial biogenesis [
73], the increased mito% in LHON carriers and cases [
12] could be explained by oxidative stress inducing larger mitochondrial volume ratios in the axon. It is possible that in carriers (i.e., before axonal degeneration occurs), the higher ROS levels signal locally or to the soma that more axonal mitochondria should be produced. Given that LHON mutations lead to higher ROS production [
23,
24], an intrinsic mitochondrial production signaling mechanism could explain the increased mito% observed in LHON cases and carriers relative to controls [
12].
This study has several limitations. First, although it is well known that cellular membranes are relatively permeable to H
2O
2 and relatively impermeability to
, and the basis for the simulations that were used, the degree of permeability to H
2O
2 also affects the results. However, it is not known what the RGC axonal permeability to H
2O
2 is in either the unmyelinated or myelinated regions, and assumptions from non-axonal cell membranes may not be incorrect. Second, although the range of concentrations at which H
2O
2 causes oxidative stress are known [
47], there is a lack of experimental data regarding the long-term effects of these concentrations on RGC axons or somas, or the time frame at which they would cause axonal degeneration. The biochemical linkage between oxidatively damaging H
2O
2 concentrations and mechanisms of axonal degeneration are also unknown.
Third, in silico studies are hampered by the balance between computational resources needed, simulation time, and simulation resolution. Our study is no exception. Model sizes and the model resolution were limited by the available GPU with its on-board memory and the need for simulation throughput. The methods presented in this paper can be extended to larger tissue samples. For example, extending the current work to a complete optic nerve and ultimately to an optic nerve-retina system would be worthwhile. Nevertheless, although the model can be extended spatially using more greater computational resources, there are limits to the simulation time that can be afforded. When a second in the biological life of the sample can only be processed in many seconds of computer simulation, longer-term evolving diseases such as LHON cannot be simulated at the time scales of the illness. On the other hand, we do not know how long the pathophysiological process takes, and are limited by patient symptoms, measurements of visual loss, clinical observation of disc edema and other retinal findings, and detection of axon and ganglion cell/inner plexiform layer loss in the retina.
Fourth, the timelines of the simulated processes are more rapid than the timeline of visual loss in LHON. Given that the simulation results are based on realistic biological parameters, the difference in timing suggests a possible intermediate mechanism triggered by ROS, which subsequently results in axonal dysfunction and degeneration, and eventually results in somal apoptosis. Such a mechanism could be a delay until the axon becomes dysfunctional, based on a biochemical, inflammatory, or other mechanisms.
Finsterer et al. [
74] reviewed journal-based reports of diseases seen in LHON patients which affect other organs. These data could support the extension of our hypothesis of ROS-induced damage in LHON patients to other organs, providing a comprehensive explanation of the observed organ problems. For example, a study by Orssaud [
75] reports abnormal cardiac function in LHON patients and recommends cardiac evaluation for such sufferers. The linkage of ROS to the most common cardiac problems, such as arrhythmia [
76], sudden cardiac death [
77], and the homoplastic nature of the LHON genetic defects, provides support for oxidative stress as a possible explanation for the misfunction of various organs in LHON patients.
No similar data regarding carriers were found in the literature. An interesting data point would be to see if non-visual diseases occur more or less frequently in carriers vs. controls or vs. LHON cases.
In summary, a more comprehensive model of ROS in LHON and the effects on the propagation of axonal degeneration has been developed. The model is more realistic anatomically, biophysically, and biochemically, and has implications for the pathophysiology of the disease and the development of therapeutic approaches. Future studies could address biochemical feedback loops between RGC and glia, the role of other ROS, the specific anatomy of the optic nerve head, and use these refinements to further advance the detection and treatment of LHON in those at risk.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












