

Multiplex Proteomic Evaluation in Inborn Errors with Deregulated IgE Response
 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Recruitment, Characterization of the Patients, and Study Design
2.2. Genomic Analysis
2.2.1. Genomic Hybridization Microarray (Array–CGH) Analysis
2.2.2. SPINK5 Molecular Analysis and LEKTI Expression
2.3. IgE Antibody Measurements
2.3.1. ImmunoCAP ISAC®
2.3.2. Allergy Explorer–ALEX2® Macroarray Analysis
3. Results
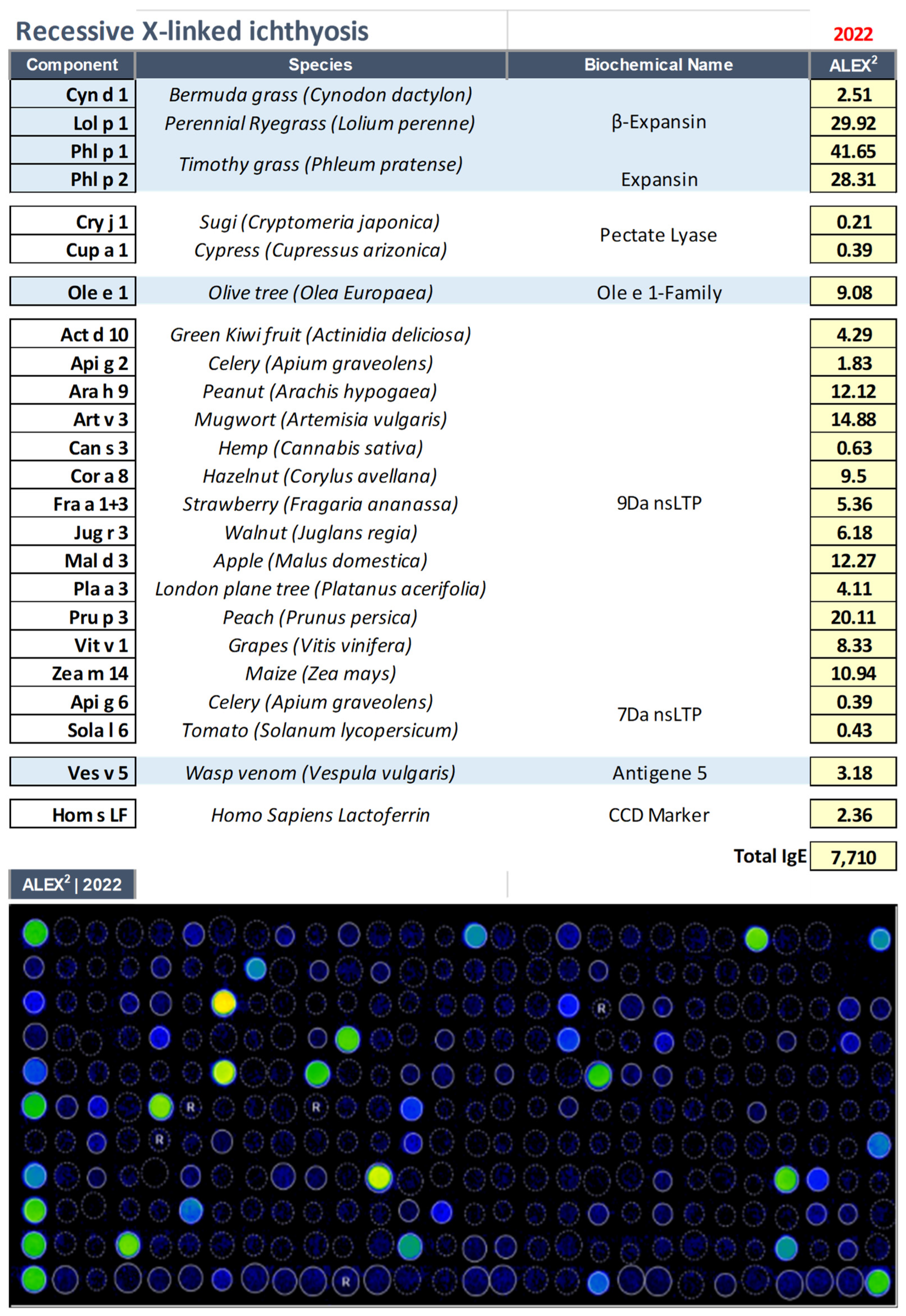
3.1. Recessive X–Linked Ichthyosis (RXLI, OMIM: 308100)
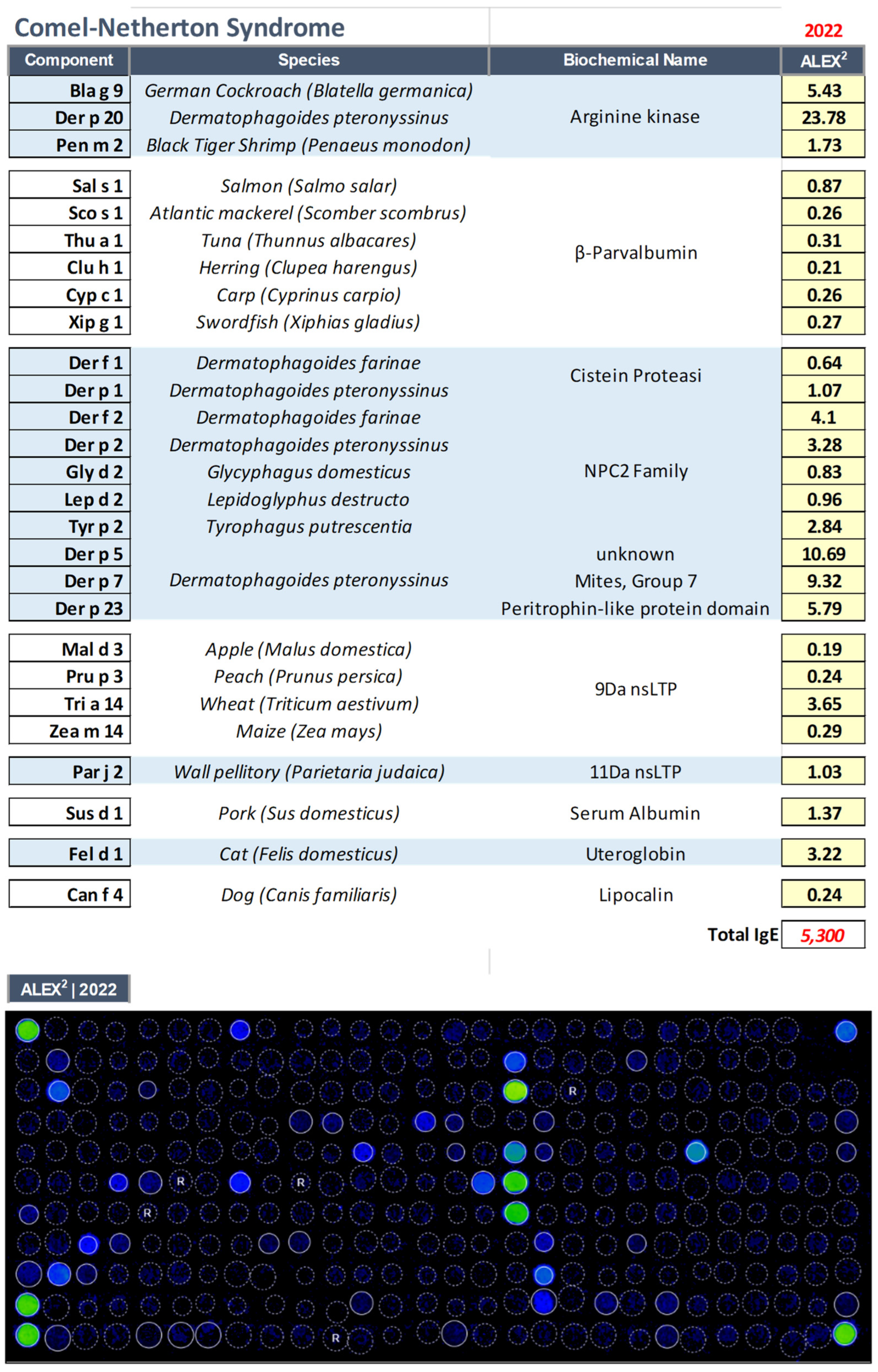
3.2. Comel–Netherton Syndrome (NS, OMIM: 256500)
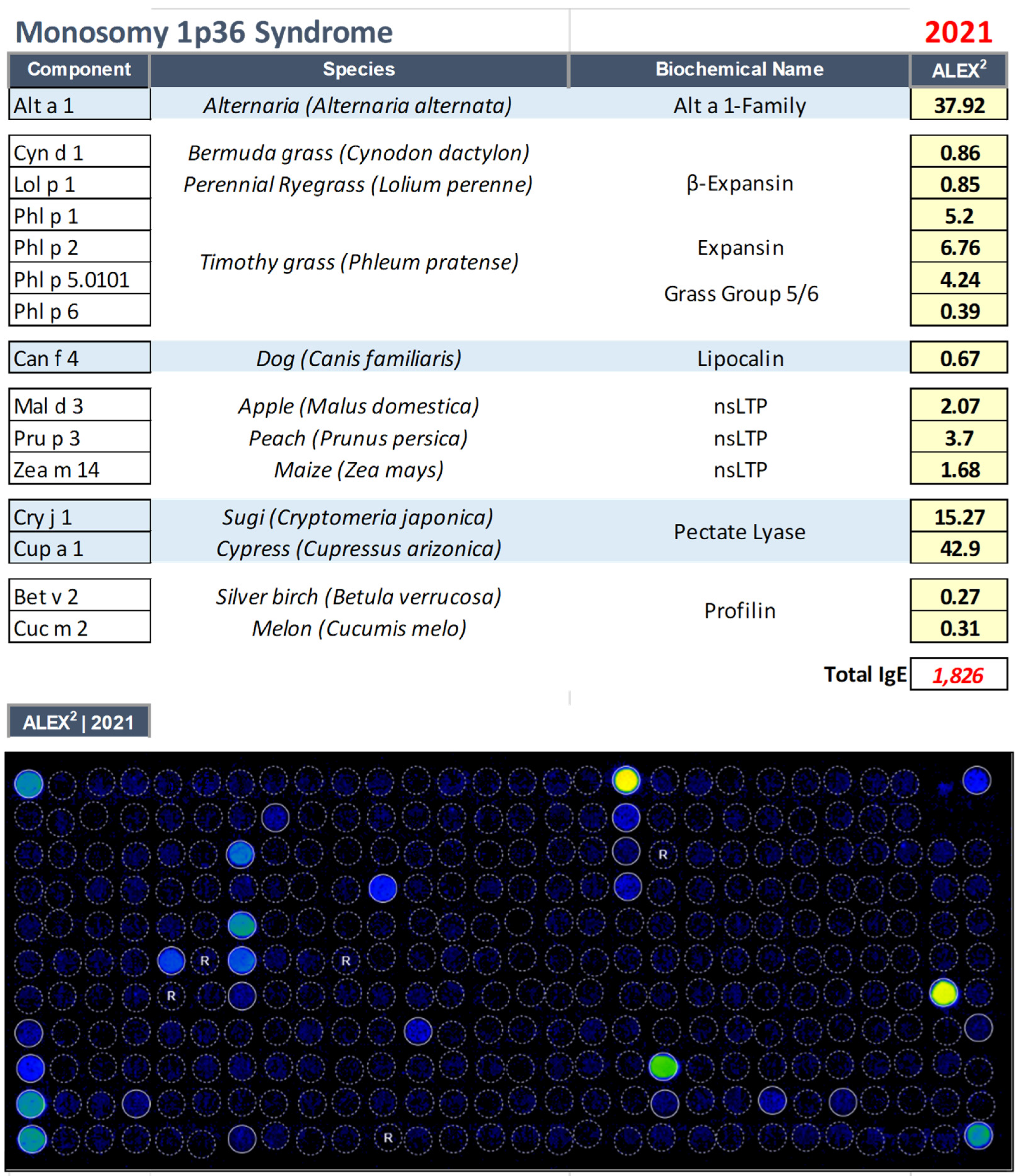
3.3. Monosomy 1p36 Syndrome (OMIM: 607872)
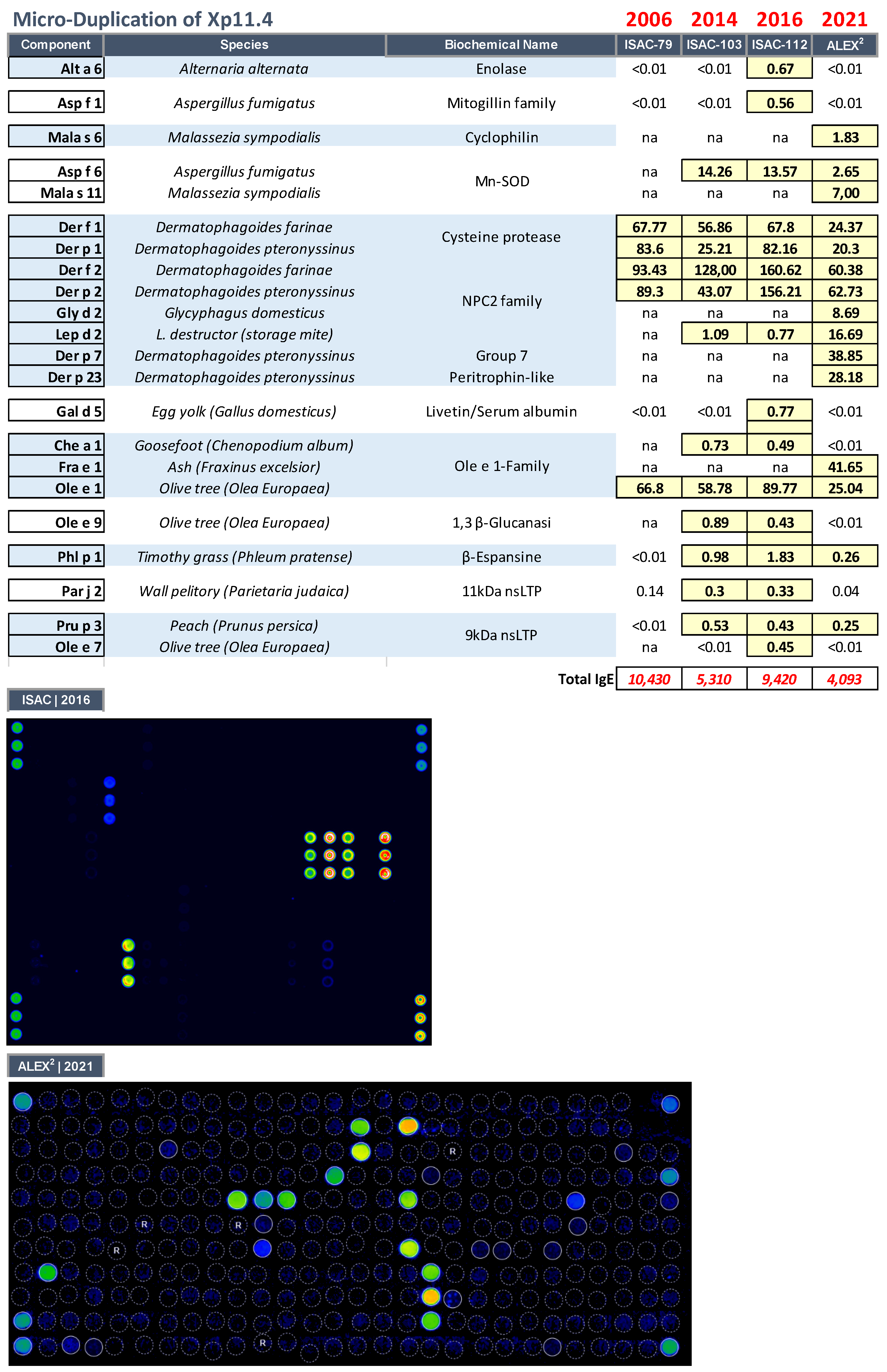
3.4. Microduplication of Xp11.4 in a Girl with a Cognitive Defect, Cerebellar Hypoplasia, Atopic Dermatitis, and Hyper–IgE Syndrome
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lobefaro, F.; Gualdi, G.; Di Nuzzo, S.; Amerio, P. Atopic Dermatitis: Clinical Aspects and Unmet Needs. Biomedicines 2022, 10, 2927. [Google Scholar] [CrossRef] [PubMed]
- Fania, L.; Moretta, G.; Antonelli, F.; Scala, E.; Abeni, D.; Albanesi, C.; Madonna, S. Multiple Roles for Cytokines in Atopic Dermatitis: From Pathogenic Mediators to Endotype–Specific Biomarkers to Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 2684. [Google Scholar] [CrossRef] [PubMed]
- Tanei, R. Clinical characteristics, treatments, and prognosis of atopic eczema in the elderly. J. Clin. Med. 2015, 4, 979–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Leonard, A.; Pavel, A.B.; Malik, K.; Raja, A.; Glickman, J.; Estrada, Y.D.; Peng, X.; del Duca, E.; Sanz–Cabanillas, J.; et al. Age–specific changes in the molecular phenotype of patients with moderate–to–severe atopic dermatitis. J. Allergy Clin. Immunol. 2019, 144, 144–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mari, A.; Scala, E.; Alessandri, C. The IgE–microarray testing in atopic dermatitis: A suitable modern tool for the immunological and clinical phenotyping of the disease. Curr. Opin. Allergy Clin. Immunol. 2011, 11, 438–444. [Google Scholar] [CrossRef]
- Muhandes, L.; Chapsa, M.; Pippel, M.; Behrendt, R.; Ge, Y.; Dahl, A.; Yi, B.; Dalpke, A.; Winkler, S.; Hiller, M.; et al. Low Threshold for Cutaneous Allergen Sensitization but No Spontaneous Dermatitis or Atopy in FLG–Deficient Mice. J. Investig. Dermatol. 2021, 141, 2611–2619.e2. [Google Scholar] [CrossRef]
- Liang, Y.; Chang, C.; Lu, Q. The Genetics and Epigenetics of Atopic Dermatitis—Filaggrin and Other Polymorphisms. Clin. Rev. Allergy Immunol. 2016, 51, 315–328. [Google Scholar] [CrossRef]
- Heo, W.I.; Park, K.Y.; Lee, M.; Bae, Y.J.; Moon, N.J.; Seo, S.J. Association of DOCK8, IL17RA, and KLK12 Polymorphisms with Atopic Dermatitis in Koreans. Ann. Dermatol. 2020, 32, 197–205. [Google Scholar] [CrossRef]
- Grimbacher, B.; Holland, S.M.; Puck, J.M. Hyper–IgE syndromes. Immunol. Rev. 2005, 203, 244–250. [Google Scholar] [CrossRef]
- Boos, A.C.; Hagl, B.; Schlesinger, A.; Halm, B.E.; Ballenberger, N.; Pinarci, M.; Heinz, V.; Kreilinger, D.; Spielberger, B.D.; Schimke–Marques, L.F.; et al. Atopic dermatitis, STAT3– and DOCK8–hyper–IgE syndromes differ in IgE–based sensitization pattern. Allergy 2014, 69, 943–953. [Google Scholar] [CrossRef]
- Briot, A.; Deraison, C.; Lacroix, M.; Bonnart, C.; Robin, A.; Besson, C.; Dubus, P.; Hovnanian, A. Kallikrein 5 induces atopic dermatitis–like lesions through PAR2–mediated thymic stromal lymphopoietin expression in Netherton syndrome. J. Exp. Med. 2009, 206, 1135–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamee, M.; Zaki–Dizaji, M.; Lo, B.; Abolhassani, H.; Aghamahdi, F.; Mosavian, M.; Nademi, Z.; Mohammadi, H.; Jadidi–Niaragh, F.; Rojas, M.; et al. Clinical, Immunological, and Genetic Features in Patients with Immune Dysregulation, Polyendocrinopathy, Enteropathy, X–linked (IPEX) and IPEX–like Syndrome. J. Allergy Clin. Immunol. Pract. 2020, 8, 2747–2760.E7. [Google Scholar] [CrossRef] [PubMed]
- Scala, E.; Condorelli, A.G.; Scala, A.; Caprini, E.; Didona, B.; Paganelli, R.; Castiglia, D. IgE Sensitization Profile in Patients with Netherton Syndrome. Int. Arch. Allergy Immunol. 2022, 183, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, D.; Scala, E.; Pomponi, D.; Caprini, E.; Arcelli, D.; Gambineri, E.; Russo, G.; Mari, A. Proteomics plus genomics approaches in primary immunodeficiency: The case of immune dysregulation, polyendocrinopathy, enteropathy, X–linked (IPEX) syndrome. Clin. Exp. Immunol. 2011, 2, 120–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scala, E.; Abeni, D.; Guerra, E.C.; Pirrotta, L.; Locanto, M.; Meneguzzi, G.; Giani, M.; Russo, G.; Asero, R. β–1,3–glucanase rOle e 9 and MnSOD rAsp f 6 IgE reactivity are the signature of atopic dermatitis in the Mediterranean area. Clin. Exp. Allergy 2020, 50, 487–498. [Google Scholar] [CrossRef]
- Raghunath, M.; Tontsidou, L.; Oji, V.; Aufenvenne, K.; Schürmeyer–Horst, F.; Jayakumar, A.; Ständer, H.; Smolle, J.; Clayman, G.L.; Traupe, H. SPINK5 and netherton syndrome: Novel mutations, demonstration of missing LEKTI, and differential expression of transglutaminases. J. Investig. Dermatol. 2004, 123, 474–483. [Google Scholar] [CrossRef] [Green Version]
- Bitoun, E.; Micheloni, A.; Lamant, L.; Bonnart, C.; Tartaglia–Polcini, A.; Cobbold, C.; Al Saati, T.; Mariotti, F.; Mazareeuw–Hautier, J.; Boralevi, F.; et al. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Hum. Mol. Genet. 2003, 12, 2417–2430. [Google Scholar] [CrossRef] [Green Version]
- Elias, P.M.; Crumrine, D.; Rassner, U.; Hachem, J.P.; Menon, G.K.; Man, W.; Choy, M.H.W.; Leypoldt, L.; Feingold, K.R.; Williams, M.L. Basis for Abnormal Desquamation and Permeability Barrier Dysfunction in RXLI. J. Investig. Dermatol. 2004, 122, 314–319. [Google Scholar] [CrossRef]
- Mari, A.; Ooievaar–De Heer, P.; Scala, E.; Giani, M.; Pirrotta, L.; Zuidmeer, L.; Bethell, D.; Van Ree, R. Evaluation by double–blind placebo–controlled oral challenge of the clinical relevance of IgE antibodies against plant glycans. Allergy 2008, 63, 891–896. [Google Scholar] [CrossRef]
- Chavanas, S.; Bodemer, C.; Rochat, A.; Hamel–Teillac, D.; Ali, M.; Irvine, A.D.; Bonafé, J.L.; Wilkinson, J.; Taïeb, A.; Barrandon, Y.; et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat. Genet. 2000, 25, 141–142. [Google Scholar] [CrossRef]
- Shapira, S.K.; McCaskill, C.; Northrup, H.; Spikes, A.S.; Elder, F.F.B.; Sutton, V.R.; Korenberg, J.R.; Greenberg, F.; Shaffer, L.G. Chromosome 1p36 deletions: The clinical phenotype and molecular characterization of a common newly delineated syndrome. Am. J. Hum. Genet. 1997, 61, 642–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquin, C.; Landais, E.; Poirsier, C.; Bénech, C.; Callier, P.; Afenjar, A.; Bednarek, N.; Bosquet, D.; Burglen, L.; Herissant, L.; et al. 1p36 deletion syndrome: Review and mapping with further characterization of the phenotype, a new cohort of 86 patients. Am. J. Med. Genet. Part A, 2022; 1–14, ahead of print. [Google Scholar] [CrossRef]
- Scala, E.; Till, S.J.; Asero, R.; Abeni, D.; Guerra, E.C.; Pirrotta, L.; Paganelli, R.; Pomponi, D.; Cecchi, L.; Giani, M.; et al. Lipid transfer protein sensitization: Reactivity profiles and clinical risk assessment in an Italian cohort. Allergy 2015, 70, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Schmid–Grendelmeier, P.; Flückiger, S.; Disch, R.; Trautmann, A.; Wüthrich, B.; Blaser, K.; Scheynius, A.; Crameri, R. IgE–mediated and T cell–mediated autoimmunity against manganese superoxide dismutase in atopic dermatitis. J. Allergy Clin. Immunol. 2005, 115, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.K.; Zaveri, H.P.; Scott, D.A. 1p36 deletion syndrome: An update. Appl. Clin. Genet. 2015, 8, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassani, S.; Cingolani, L.A.; Valnegri, P.; Folci, A.; Zapata, J.; Gianfelice, A.; Sala, C.; Goda, Y.; Passafaro, M. The X–Linked Intellectual Disability Protein TSPAN7 Regulates Excitatory Synapse Development and AMPAR Trafficking. Neuron 2012, 73, 1143–1158. [Google Scholar] [CrossRef] [Green Version]
- Palka, C.; De Marco, S.; Alfonsi, M.; Matricardi, S.; Errichiello, E.; Morizio, E.; Franchi, P.G.; Calabrese, G.; Mohn, A.; Chiarelli, F. Discovering a familial Xp11.4 microduplication: Does the mother matter? Meta Gene 2018, 16, 90–95. [Google Scholar] [CrossRef]
- Tarrant, J.M.; Robb, L.; Van Spriel, A.B.; Wright, M.D. Tetraspanins: Molecular organisers of the leukocyte surface. Trends Immunol. 2003, 24, 610–617. [Google Scholar] [CrossRef]
- Levy, S.; Shoham, T. The tetraspanin web modulates immune–signalling complexes. Nat. Rev. Immunol. 2005, 5, 136–148. [Google Scholar] [CrossRef]
- Van Spriel, A.B. Tetraspanins in the humoral immune response. Biochem. Soc. Trans. 2011, 39, 512–517. [Google Scholar] [CrossRef]
- Peng, W.M.; Yu, C.F.; Kolanus, W.; Mazzocca, A.; Bieber, T.; Kraft, S.; Novak, N. Tetraspanins CD9 and CD81 are molecular partners of trimeric FcεRI on human antigen–presenting cells. Allergy 2011, 66, 605–611. [Google Scholar] [CrossRef]
- Maecker, H.T.; Do, M.S.; Levy, S. CD81 on B cells promotes interleukin 4 secretion and antibody production during T helper type 2 immune responses. Proc. Natl. Acad. Sci. USA 1998, 95, 2458–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, P.S.; Maltby, S.; Rosenberg, H.F.; Tay, H.L.; Hogan, S.P.; Collison, A.M.; Yang, M.; Kaiko, G.E.; Hansbro, P.M.; Kumar, R.K.; et al. Modeling TH2 responses and airway inflammation to understand fundamental mechanisms regulating the pathogenesis of asthma. Immunol. Rev. 2017, 278, 20–40. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.F. X–chromosome inactivation and developmental patterns in mammals. Biol. Rev. Camb. Philos. Soc. 1972, 47, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Della Ragione, F.; Scalabrì, F.; D’Esposito, M. X inactivation and reactivation in X–linked diseases. Semin. Cell Dev. Biol. 2016, 56, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Monnot, S.; Giuliano, F.; Massol, C.; Fossoud, C.; Cossée, M.; Lambert, J.C.; Karmous–Benailly, H. Partial Xp11.23–p11.4 duplication with random X inactivation: Clinical report and molecular cytogenetic characterization. Am. J. Med. Genet. Part A 2008, 146, 1325–1329. [Google Scholar] [CrossRef]
- Hulst, M.; Jansman, A.; Wijers, I.; Hoekman, A.; Vastenhouw, S.; van Krimpen, M.; Smits, M.; Schokker, D. Enrichment of in vivo transcription data from dietary intervention studies with in vitro data provides improved insight into gene regulation mechanisms in the intestinal mucosa. Genes Nutr. 2017, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- El–Sayed, Z.A.; El–Ghoneimy, D.H.; Ortega–Martell, J.A.; Radwan, N.; Aldave, J.C.; Al–Herz, W.; Al–Nesf, M.A.; Condino–Neto, A.; Cole, T.; Eley, B.; et al. Allergic Manifestations of Inborn Errors of Immunity and Their Impact on the Diagnosis: A Worldwide Study. World Allergy Organ. J. 2022, 15, 100657. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Sex | Disease |
|---|---|---|
| 12 yrs | Male | Recessive X–linked ichthyosis (RXLI, OMIM: 308100) |
| 52 yrs | Male | Comel–Netherton Syndrome (NS, OMIM: 256500) |
| 20 yrs | Female | Monosomy 1p36 Syndrome (OMIM: 607872) |
| 7 yrs | Female | Microduplication of Xp11. |
 | ||||||||
| Cytogenetic location | Genomic coordinates (NCBI/GRCh38) | Gene Locus | Gene Locus name | Gene Locus MIM number | Approved Symbol | Entrez Gene ID | Phenotype | Inheritance |
| Xp11.4 | X:38561541-38688917 | TSPAN7, TM4SF2, MXS1, A15, XLID58 | Tetraspanin 7 | 300096 | TSPAN7 | 7102 | Intellectual developmental disorder, X-linked 58 | X-linked recessive |
| Xp11.4 | X:38801458-38806531 | MID1IP1, MIG12 | MID1-interacting protein 1 | 300961 | MID1IP1 | 58526 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scala, E.; Madonna, S.; Castiglia, D.; Scala, A.; Caprini, E.; Paganelli, R. Multiplex Proteomic Evaluation in Inborn Errors with Deregulated IgE Response. Biomedicines 2023, 11, 202. https://doi.org/10.3390/biomedicines11010202
Scala E, Madonna S, Castiglia D, Scala A, Caprini E, Paganelli R. Multiplex Proteomic Evaluation in Inborn Errors with Deregulated IgE Response. Biomedicines. 2023; 11(1):202. https://doi.org/10.3390/biomedicines11010202
Chicago/Turabian StyleScala, Enrico, Stefania Madonna, Daniele Castiglia, Alessandro Scala, Elisabetta Caprini, and Roberto Paganelli. 2023. "Multiplex Proteomic Evaluation in Inborn Errors with Deregulated IgE Response" Biomedicines 11, no. 1: 202. https://doi.org/10.3390/biomedicines11010202
APA StyleScala, E., Madonna, S., Castiglia, D., Scala, A., Caprini, E., & Paganelli, R. (2023). Multiplex Proteomic Evaluation in Inborn Errors with Deregulated IgE Response. Biomedicines, 11(1), 202. https://doi.org/10.3390/biomedicines11010202