1. Introduction
The contagious disease called coronavirus disease 2019 (COVID-19) is brought on by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [
1]. The virus, which was first discovered in Wuhan in December 2019, has since spread around the world, causing the deaths of millions and having catastrophic effects on the economy and society. The necessity for effective vaccinations is therefore extremely important [
2]. Several strategies have been put up by researchers to create SARS-CoV-2 vaccines [
3,
4]. Growing pathogens is the basis of the conventional vaccine development method, which makes it exceedingly time-consuming to isolate, inactivate, and inject the disease-causing virus [
5]. This method typically takes over a year to produce effective vaccinations, and as a result, it does very little to stop the disease’s spread [
6,
7].
The current COVID-19 control measures, which do not yet include a single particular antiviral drug for SARS-CoV-2, include early diagnosis, reporting, isolation, and supportive therapies [
8]. Elderly persons and those with weakened immune systems tend to have more severe illnesses and are more likely to perish because their cells’ ability to resist infection and repair themselves is decreased by a compromised immune system [
9]. The immune system has two distinct response mechanisms: innate and adaptive. Innate immunity swiftly intervenes and initiates early immune responses when it comes into contact with pathogens [
10]. On the other hand, adaptive immunity may be developed in several ways, including through disease exposure or externally delivered serum and vaccinations. Adaptive immune systems contain memories that allow them to recall previous infections; as such, antigen-specific responses are produced by adaptive immune systems [
11]. Adaptive immune responses come in both humoral and cellular forms. A humoral response utilizes B-cells to produce antibodies that can target an antigen when exposed to it. To create a vaccine, the highly immunogenic portions of the protein of a pathogenic organism must first be identified. These areas are referred to as B- and T-cell epitopes and are in charge of triggering immune responses [
12,
13].
SARS-CoV-2 has a large 26–32 kb RNA genome that encodes several structural and non-structural proteins, such as Spike (S), Envelope (E), Membrane (M), and Nucleocapsid (N), which are crucial for triggering immunological responses [
14,
15,
16,
17]. Therefore, an epitope peptide vaccine made of viral proteins S, M, N, and E is highly required to control disease spread and another SARS virus in the future. B-cell epitopes can be linear or conformational. While conformational epitopes are made up of amino acids that are connected during the folding of a protein, linear epitopes are produced by a sequence of the amino acids of the protein [
13,
18]. One study [
19] identified the best epitopes for an epitopic vaccination to prevent SARS-CoV-2 infection. To identify and describe potential B and T-cell epitopes for the creation of the epitopic vaccine, immunoinformatics were used. The SARS-CoV-2 spike glycoprotein was selected as the target because it creates the virus’ distinctive crown and protrudes from the viral membrane. Multiple servers and pieces of software built on the immunoinformatic platform were used to explore the spike glycoprotein’s protein sequence. The focus of this study is on linear epitope prediction due to the limited number of available datasets for conformational epitopes. Conventional approaches for creating vaccines against deadly diseases have proven to be exceedingly time- and money-consuming. Using in silico techniques, vaccine candidates for earlier viruses (Zika, Ebola, HPV, and MERS) have been successfully designed [
20,
21].
In a study, immunoinformatics-based techniques were used to find possible immunodominant SARS-CoV-2 epitopes, which may be relevant to creating COVID-19 vaccines. In total, 25 epitopes that were 100% similar to experimentally verified SARS-CoV epitopes and 15 putative immunogenic areas from three SARS-CoV-2 proteins were found. Analysis was carried out to test the suitability of the epitopes as a vaccine [
22]. Similarly, immunoinformatics tools were used to create a multi-epitope vaccine that could be employed for COVID-19 prevention as well as treatment. B-cell, CTL, and HTL epitopes were combined to create this multi-epitope vaccine. Using online tools, additional research was done to predict and evaluate the vaccine structure and efficacy [
23]. To provide a list of possibly immunogenic and antigenic peptide epitopes that might aid in vaccine creation, several immunoinformatics methods were integrated. Spike proteins’ S1 and S2 domains were examined, and two vaccine constructions, with T- and B-cell epitopes, were given priority. Using linkers and adjuvants, prioritized epitopes were then modeled, and corresponding 3D models were built to assess their physiochemical characteristics and potential interactions with ACE2 and HLA superfamily alleles [
24].
Machine learning algorithms’ architecture automatically identifies patterns in data, which is perfect for data-driven sciences, such as genomics [
25,
26]. The usage of DL frameworks in medical imaging has been widespread [
27,
28,
29,
30,
31]. To create computer models that can more accurately predict the existence of linear B-cell epitopes from an amino acid sequence for vaccine production against a pathogenic organism, in silico techniques have been frequently employed [
32]. There are several programs available and cited in the literature that employ machine learning methods to predict linear B-cell epitopes, including BepiPred-2.0 [
33], BCPred [
34], EpiDope [
35], ABCPred [
36], and SVMTrip [
37]. In addition, the Lbtope tool and SVM, K-nearest neighbor models [
38], and genetic algorithms [
39] have been used in vaccine design for B-cell or T-cell epitopes. Most of these machine learning models rely on features related to amino acid sequences; therefore, the portrayed effectiveness of such models is ineffective.
The web server BepiPred-2.0 is used to predict B-cell epitopes from antigen sequences. The data utilized had 11,834 positive and 18,722 negative epitopes, and it was taken from the immune epitope database (IEDB) [
40]. Since epitopes are rarely found outside of peptides consisting of five to twenty-five amino acids, the peptides within this range were eliminated. The random forest (RF) regression technique with fivefold cross-validation was utilized as the training approach; the method was reported to have an AUC of 0.62 on test datasets [
33]. BCPred, another method to predict linear B-cell epitopes, employed SVM that utilized a radial-based kernel with five kernel modifications and fivefold cross-validation. The proposed technique eventually achieved an AUC of around 0.76 [
34]. In another study, deep neural networks were used by the application EpiDope to locate B-cell epitopes in specific protein sequences composed of ELMo DNN and biLSTM. Each of the 30,556 protein sequences in the dataset, which was taken from the IEDB, had experimentally validated epitopes or non-epitopes. A bidirectional LSTM (long short-term memory) layer was linked to a vector of length 10 that was used to encode each amino acid in the ELMo DNN branch. Tenfold cross-validation was employed for validation, achieving an AUC of 0.67 [
35]. ABCPred employed a recurrent neural network (RNN) to predict linear B-cell epitopes. For each residue, sliding windows containing 10 to 20 amino acids were employed to determine its attributes. Fivefold cross-validation was utilized for these tests, yielding an accuracy of 66% [
36]. The SVMTriP approach, which combines SVM with tri-peptide similarity and propensity scores, is used to predict linear antigenic B-cell epitopes. Our dataset was taken from the IEDB and consisted of 65,456 positive epitopes [
40]. Using fivefold cross-validation, it obtained an AUC value of 0.702. The authors of [
41] created a strategy for predicting B-cell linear epitopes that was based on the design of a fuzzy-ARTMAP neural network. This was trained on 15 attributes, including an amino acid ratio scale and a set of 14 physicochemical scales, using a linear averaging approach. Fivefold cross-validation procedures were employed with datasets taken from the IEDB to train and validate the knowledge of models and were shown to achieve an AUC of 0.7831 on test data, which is a good performance. To take into account the properties of a whole antigen protein in combination with the target sequence, [
42] presents a deep learning approach based on short-term memory with an attention mechanism. In experimental epitope location prediction with data taken from the immune epitope database, the suggested technique outperformed the standard method, with an AUC of 0.822. The SMOTE technique produced more accurate predictions than other methods when evaluated for the datasets balanced using it. It was shown that once the SARS-CoV and B-cell datasets used for training were balanced, the epitope prediction success of the models generally rose, with an accuracy of 0.914.
There are very few deep learning methods available for forecasting B-cell epitopes, and even though some of the models are good at predicting linear B-cell epitopes, based on their performances, they still have some difficulties with making good predictions, which makes it necessary to develop better models. This research suggests a novel and effective hybrid transfer learning strategy, for forecasting SARS-CoV-2 epitopes, that integrates data preprocessing of physiochemical characteristics and sequence-based features. This study’s goal is to provide a better method of identifying the epitope areas that could be candidates for vaccines. The list of abbreviations is presented in
Table 1. The contributions made by this study are as follows:
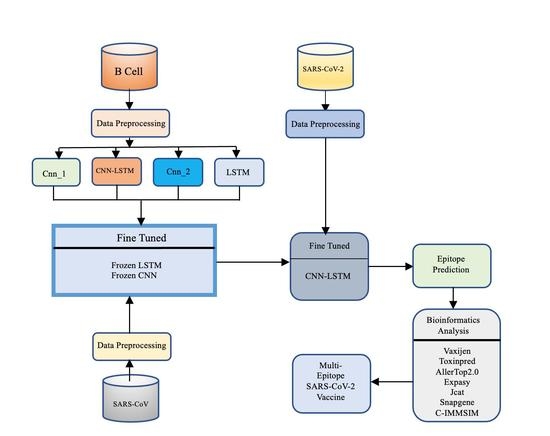
We proposed CNN-LSTM, a hybrid architecture that combines a CNN with bidirectional LSTM (BiLSTM), to predict B-cell epitopes given a peptide sequence that may be employed for vaccine development. The idea behind this hybrid architecture is to employ the CNN for feature extraction and LSTM for modeling feature relationships in order of appearance.
To address the issue of limited sample sizes and enhance model performance, we put forth a transfer learning technique. In particular, we first pre-trained the proposed CNN-LSTM using the B-cell dataset. The pre-trained CNN-LSTM was then adjusted to predict epitopes using SARS-CoV datasets.
To forecast epitopes from the SARS-CoV-2 datasets, we looked into three transfer learning strategies—fine-tuned, frozen CNN, and frozen LSTM—using information learnt from the previous model.
With the use of the bioinformatics tools AllerTop, VaxiJen, ToxinPred, Jcat, and Snapgene, we investigated epitopes discovered with transfer learning techniques.
Table 1.
List of abbreviations.
Table 1.
List of abbreviations.
| Abbreviation | Meaning |
|---|
| AUC | Area under the curve |
| CAI | Codon optimization index |
| CNN | Convolutional neural network |
| COVID-19 | Coronavirus disease 2019 |
| DL | Deep learning |
| IEDB | Immune epitope database |
| IgM and IgG | Immunoglobulins IgM and IgG |
| IFN-γ | Interferon |
| JCat | Java Codon Adaptation Tool |
| LSTM | Long short-term memory |
| ReLU | Rectified Linear Unit |
| RF | Random forest |
| RNN | Recurrent neural network |
| ROC | Receiver operating characteristic |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| Tc | Cytotoxic T lymphocytes |
| Th | Helper T lymphocytes |
4. Discussion
In this work, a CNN-LSTM method was pre-trained using a B-cell dataset; the pre-trained CNN-LSTM was then adjusted using SARS-CoV datasets, and that knowledge was transferred to a new, fine-tuned CNN-LSTM model. It could be seen that combining the CNN with LSTM was a good strategy, since there was an increase in performance from using either the CNN or LSTM alone. The first CNN-LSTM model was trained using the B-cell dataset. In the second stage of the knowledge transfer technique, the model was fine-tuned using SARS-CoV datasets. It could be observed that there was an 18% improvement in the accuracy of the model. There are a limited number of machine learning tools for epitope determination. Additionally, the accuracy of some methods, when compared to our CNN-LSTM performance for the epitope prediction task, is low; our method gave a better performance even when compared to the best method so far. These methods were recently developed using the same datasets utilized in this study, so they were chosen for fair comparison in the prediction of SARS-CoV epitopes. The ensemble utilized by [
62] was a layered ensemble, with XGBoost on the outer layer and random forest regression with gradient boosting at the inner layer, and gave an AUC of 0.923 and an accuracy of 87.79%. In another study [
42], the attention method and LSTM were paired for this task by combining protein with chemical and structural features, during which they achieved an accuracy of 79% and an AUC of 0.822. In another study, a Bayesian neural network was used for this task and was able to achieve an accuracy of 85% [
63]. It can be seen from
Table 9 that the random forest model developed in [
43] was able to make good predictions, although there is a need for improvement in the model’s performance. In all the methods used previously, to the best of our knowledge, none have attempted the transfer learning strategy that was used in this study. This technique was able to significantly improve performance to an accuracy of 95.1% and an AUC of 0.979, which are better than all the results obtained in other methods. In this study, the performance of the proposed method was compared with that of state-of-the-art methods, and it gave outstanding results, with 95.1% accuracy. The benchmark deep learning model was an attention–LSTM-based model and was able to achieve 79% accuracy. Therefore, there was about a 16% increase in performance.
In the next stage, we tried to analyze the predicted epitopes to screen the best ones for vaccine development. Here, tools such as Toxinpred were utilized for determination of toxicity. When a peptide sequence is provided to the tool, it will return a score, and when that score is less than 0.0, then the peptide sequence is non-toxic. Other parameters, such as molecular weight and hydrophilicity, can be seen with the tool. It has been found that peptides with lower molecular weights tend to be non-toxic [
52], and hydrophilicity is quite important as well, for generating the process of the immune response [
64]. A good vaccine must not cause allergies; hence, the prediction of vaccine allergenicity is crucial. We used the AllerTOP2.0 service, which assesses peptide allergenicity. Among the predicted epitopes, twelve were considered non-allergens, while four were allergens, according to the AllerTOP2.0 [
53]. Finally, to choose the best sequence for vaccine development, it is important to determine antigenicity because antigenic peptides will generate high immune responses in humans. For this task, the VaxiJen tool was used to predict antigenicity [
54].
In order to check the expression of the vaccine sequence, a codon adaptation tool, Jcat, was used to optimize the codon usage of the designed vaccine. A codon optimization index (CAI) value of 1.0 was obtained, which means that the vaccine would be highly expressed in E. coli. It is important to have the vaccine expressed in the host organism in other to elicit an immune response. It was successfully cloned into the pCC1BAC vector using the snapgene tool. Further analysis with the Expasy tool revealed that the vaccine would have a half-life of 30 h in mammalian reticulocytes and would be stable. Finally, the immune response simulation showed that the vaccine sequence, when injected, would produce high numbers of B-cells and T-cells as well as a high amount of IFN-γ, which would assist other immune cells in killing the virus.
Even though fine-tuned CNN-LSTM has enhanced epitope prediction performance and has been turned into a powerful method, there are still several interesting directions to explore. Three issues will be the focus of our next approach. First, investigating more deep-learning-based frameworks and utilizing techniques for the best hyperparameter search may result in improved performance. Secondly, it is important to consider peptide sequence information in the feature space apart from the length that was considered in this work. Finally, we will explore explainable AI to understand the features that are utilized by this model for decision-making. This study will assist scientists in designing a suitable vaccine for COVID-19, which can be validated experimentally, in a short period.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






