

Anticancer Activity of Anti-Tubercular Compound(s) Designed on Pyrrolyl Benzohydrazine Scaffolds: A Repurposing Study

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
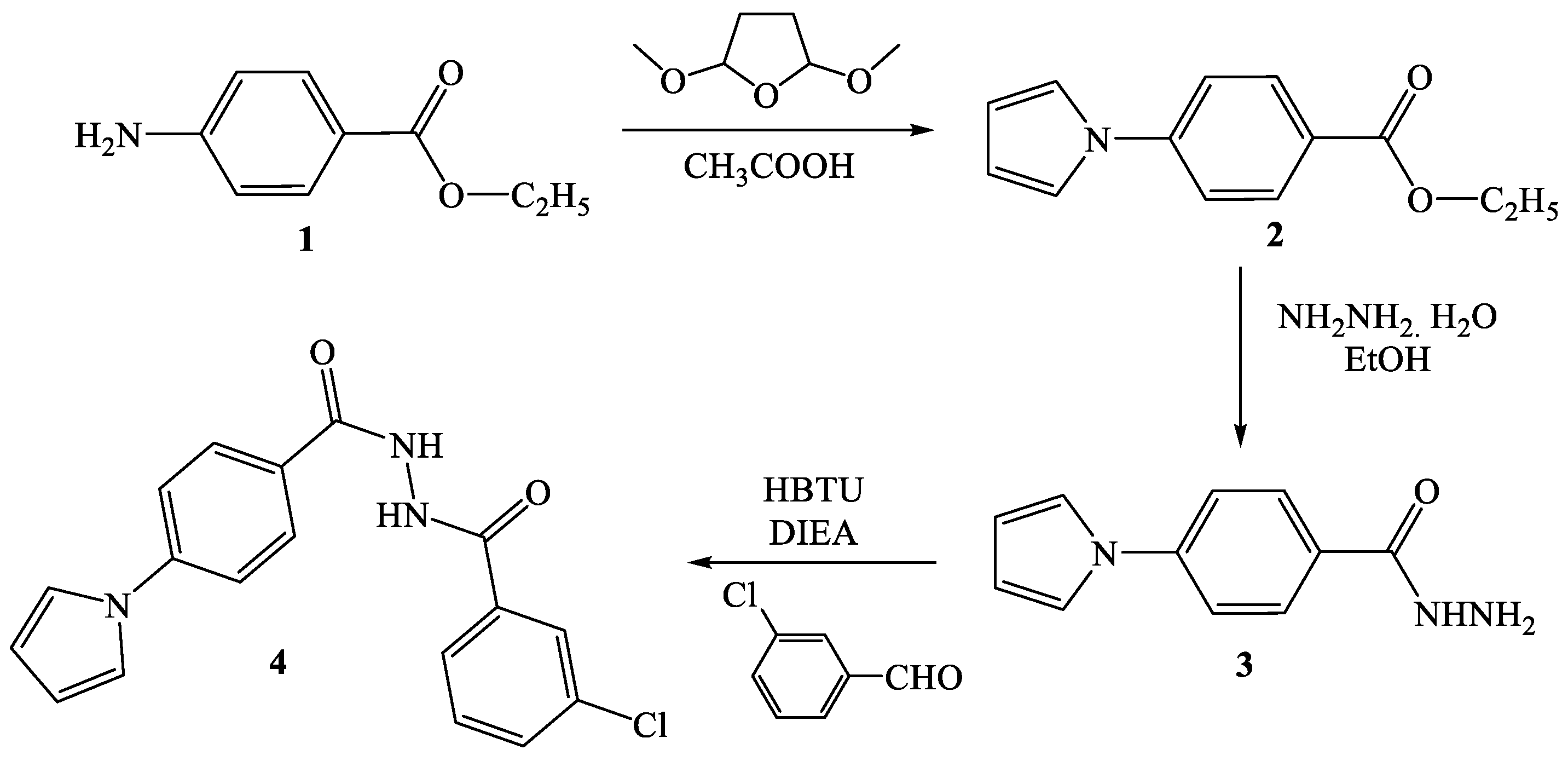
2.1. Synthesis of Pyrrolyl Benzohydrazide Derivatives
2.2. Dry Lab Investigations
2.2.1. Ligand Preparation for Molecular Docking
2.2.2. Target Preparation for Molecular Docking
2.2.3. Molecular Docking Interaction Study
2.2.4. Molecular Dynamics (MD) Simulation Study
2.3. Wet Lab Investigations
2.3.1. Materials
2.3.2. Cell Culture
2.3.3. Stock Solution of Tested Compounds
2.3.4. Cytotoxic In Vitro MTT Assay
2.3.5. Cell Cycle Analysis
2.3.6. Cell Apoptosis Assay
2.3.7. DAPI Assay
2.3.8. Statistical Analysis
3. Results and Discussion
3.1. Molecular Interaction of Pyrrolyl Benzohydrazide Derivatives with PLK1
3.2. Molecular Dynamic (MD) Analysis of C8 and C18 with PLK1
3.3. Cytotoxicity Analysis
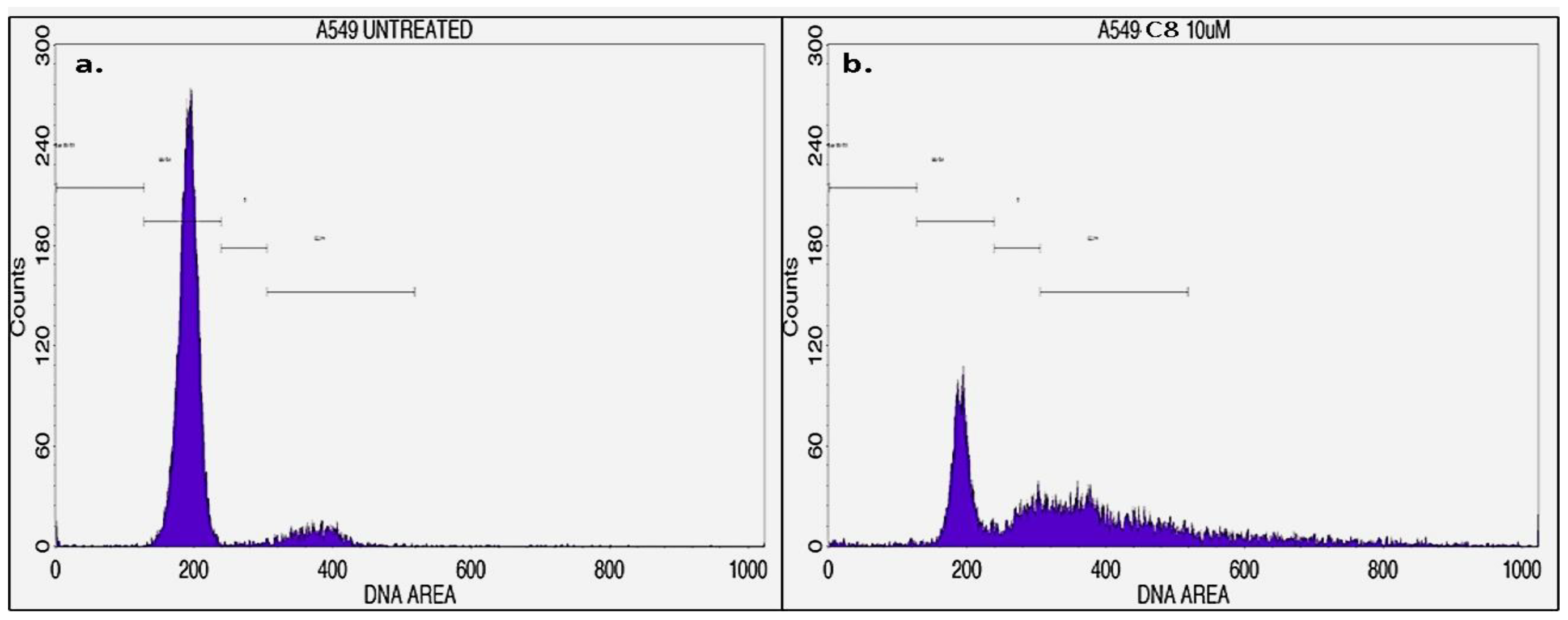
3.4. Cell Cycle Analysis
3.5. Cell Apoptosis Analysis
3.6. DAPI Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Albrecht, B.; Andersen, S.; Chauhan, K.; Graybosch, D.; Menu, P. Pursuing Breakthroughs in Cancer-Drug Development. Available online: https://www.mckinsey.com/industries/life-sciences/our-insights/pursuing-breakthroughs-in-cancer-drug-development (accessed on 25 January 2023).
- Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 25 January 2023).
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA. Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Schein, C.H. Repurposing Approved Drugs for Cancer Therapy. Br. Med. Bull. 2021, 137, 13–27. [Google Scholar] [CrossRef]
- Rodrigues, R.; Duarte, D.; Vale, N. Drug repurposing in cancer therapy: Influence of patient’s genetic background in breast cancer treatment. Int. J. Mol. Sci. 2022, 23, 4280. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.M.; Sawant, S.S.; Kunda, N.K. Inhalable Bedaquiline-Loaded Cubosomes for the Treatment of Non-Small Cell Lung Cancer (NSCLC). Int. J. Pharm. 2021, 607, 121046. [Google Scholar] [CrossRef] [PubMed]
- Parvathaneni, V.; Elbatanony, R.S.; Goyal, M.; Chavan, T.; Vega, N.; Kolluru, S.; Muth, A.; Gupta, V.; Kunda, N.K. Repurposing Bedaquiline for Effective Non-Small Cell Lung Cancer (NSCLC) Therapy as Inhalable Cyclodextrin-Based Molecular Inclusion Complexes. Int. J. Mol. Sci. 2021, 22, 4783. [Google Scholar] [CrossRef]
- Choi, J.; Park, S.-J.; Jee, J.-G. Analogues of Ethionamide, a Drug Used for Multidrug-Resistant Tuberculosis, Exhibit Potent Inhibition of Tyrosinase. Eur. J. Med. Chem. 2015, 106, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Jee, J.-G. Repositioning of Thiourea-Containing Drugs as Tyrosinase Inhibitors. Int. J. Mol. Sci. 2015, 16, 28534–28548. [Google Scholar] [CrossRef] [Green Version]
- Falzon, D.; Hill, G.; Pal, S.N.; Suwankesawong, W.; Jaramillo, E. Pharmacovigilance and Tuberculosis: Applying the Lessons of Thioacetazone. Bull. World Health Organ. 2014, 92, 918–919. [Google Scholar] [CrossRef]
- Lv, Q.; Wang, D.; Yang, Z.; Yang, J.; Zhang, R.; Yang, X.; Wang, M.; Wang, Y. Repurposing Antitubercular Agent Isoniazid for Treatment of Prostate Cancer. Biomater. Sci. 2019, 7, 296–306. [Google Scholar] [CrossRef]
- Joshi, S.D.; Dixit, S.R.; Kulkarni, V.H.; Lherbet, C.; Nadagouda, M.N.; Aminabhavi, T.M. Synthesis, Biological Evaluation and in Silico Molecular Modeling of Pyrrolyl Benzohydrazide Derivatives as Enoyl ACP Reductase Inhibitors. Eur. J. Med. Chem. 2017, 126, 286–297. [Google Scholar] [CrossRef]
- Wang, H.C.; Yan, X.Q.; Yan, T.L.; Li, H.X.; Wang, Z.C.; Zhu, H.L. Design, synthesis and biological evaluation of benzohydrazide derivatives containing dihydropyrazoles as potential EGFR kinase inhibitors. Molecules. 2016, 21, 1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basha, N.J.; Basavarajaiah, S.M.; Shyamsunder, K. Therapeutic Potential of Pyrrole and Pyrrolidine Analogs: An Update. Mol. Divers. 2022, 26, 2915–2937. [Google Scholar] [CrossRef]
- Mateev, E.; Georgieva, M.; Zlatkov, A. Pyrrole as an Important Scaffold of Anticancer Drugs: Recent Advances. J. Pharm. Pharm. Sci. 2022, 25, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Tarzia, G.; Duranti, A.; Tontini, A.; Spadoni, G.; Mor, M.; Rivara, S.; Plazzi, P.V.; Kathuria, S.; Piomelli, D. Synthesis and Structure–Activity Relationships of a Series of Pyrrole Cannabinoid Receptor Agonists. Bioorg. Med. Chem. 2003, 11, 3965–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, M.; Kumar, R. Medicinal Attributes of Pyrazolo[3,4-d]Pyrimidines: A Review. Bioorg. Med. Chem. 2013, 21, 5657–5668. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman, S. Review: Biologically Active Pyrazole Derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Saleh, N.M.; El-Gazzar, M.G.; Aly, H.M.; Othman, R.A. Novel Anticancer Fused Pyrazole Deriv Biologically active pyrazole derivatives atives as EGFR and VEGFR-2 Dual TK Inhibitors. Front. Chem. 2020, 7, 917. [Google Scholar] [CrossRef]
- Arjun, H.A.; Elancheran, R.; Manikandan, N.; Lakshmithendral, K.; Ramanathan, M.; Bhattacharjee, A.; Lokanath, N.K.; Kabilan, S. Design, Synthesis, and Biological Evaluation of (E)-N′-((1-Chloro-3,4-Dihydronaphthalen-2-Yl)Methylene)Benzohydrazide Derivatives as Anti-Prostate Cancer Agents. Front. Chem. 2019, 7, 474. [Google Scholar] [CrossRef] [Green Version]
- Morcoss, M.; Abdelhafez, E.S.; Abdel-Rahman, H.; Abdel-Aziz, M.; El-Ella, D.A. Novel Benzimidazole/Hydrazone Derivatives as Promising Anticancer Lead Compounds: Design, Synthesis, and Molecular Docking Study. J. Adv. Biomed. Pharm. Sci. 2020, 3, 45–52. [Google Scholar] [CrossRef]
- Sarno, F.; Papulino, C.; Franci, G.; Andersen, J.H.; Cautain, B.; Melardo, C.; Altucci, L.; Nebbioso, A. 3-Chloro-N′-(2-Hydroxybenzylidene) Benzohydrazide: An LSD1-Selective Inhibitor and Iron-Chelating Agent for Anticancer Therapy. Front. Pharmacol. 2018, 9, 1006. [Google Scholar] [CrossRef] [Green Version]
- Ohira, M.; Iwasaki, Y.; Tanaka, C.; Kuroki, M.; Matsuo, N.; Kitamura, T.; Yukuhiro, M.; Morimoto, H.; Pang, N.; Liu, B.; et al. A Novel Anti-Microtubule Agent with Carbazole and Benzohydrazide Structures Suppresses Tumor Cell Growth in Vivo. Biochim. Biophys. Acta-Gen. Subj. 2015, 1850, 1676–1684. [Google Scholar] [CrossRef]
- Kamath, P.R.; Sunil, D.; Ajees, A.A.; Pai, K.S.R.; Biswas, S. N′-((2-(6-Bromo-2-Oxo-2H-Chromen-3-Yl)-1H-Indol-3-Yl)Methylene)Benzohydrazide as a Probable Bcl-2/Bcl-XL Inhibitor with Apoptotic and Anti-Metastatic Potential. Eur. J. Med. Chem. 2016, 120, 134–147. [Google Scholar] [CrossRef]
- Radwan, A.A.; Al-Mohanna, F.; Alanazi, F.K.; Manogaran, P.S.; Al-Dhfyan, A. Target β-Catenin/CD44/Nanog Axis in Colon Cancer Cells by Certain N′-(2-Oxoindolin-3-Ylidene)-2-(Benzyloxy)Benzohydrazides. Bioorg. Med. Chem. Lett. 2016, 26, 1664–1670. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.M.D.; Shakil, S.; Haneef, M. A Simple Click by Click Protocol to Perform Docking: AutoDock 4.2 Made Easy for Non-Bioinformaticians. EXCLI J. 2013, 12, 831–857. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, S.M.D.; Alshammari, A.A.A.; Almawkaa, W.A.; Ahmed, A.B.F.; Katamesh, A.; Alafnan, A.; Almutairi, T.J.; Alshammari, R.F. An Oncoinformatics Study to Predict the Inhibitory Potential of Recent FDA-Approved Anti-Cancer Drugs against Human Polo-like Kinase 1 Enzyme: A Step towards Dual-Target Cancer Medication. 3 Biotech 2019, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A. 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Mandal, S.P.; Garg, A.; Prabitha, P.; Wadhwani, A.D.; Adhikary, L.; Kumar, B.R.P. Novel Glitazones as PPARγ Agonists: Molecular Design, Synthesis, Glucose Uptake Activity and 3D QSAR Studies. Chem. Cent. J. 2018, 12, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murthy, S.S.; Narsaiah, T.B. Cytotoxic Effect of Bromelain on HepG2 Hepatocellular Carcinoma Cell Line. Appl. Biochem. Biotechnol. 2021, 193, 1873–1897. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Chojnowski, G.; Waterhouse, N.J. Measuring the DNA content of cells in apoptosis and at different cell-cycle stages by propidium iodide staining and flow cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb-rot087247. [Google Scholar] [CrossRef] [PubMed]
- Andree, H.A.; Reutelingsperger, C.P.; Hauptmann, R.; Hemker, H.C.; Hermens, W.T.; Willems, G.M. Binding of Vascular Anticoagulant Alpha (VAC Alpha) to Planar Phospholipid Bilayers. J. Biol. Chem. 1990, 265, 4923–4928. [Google Scholar] [CrossRef]
- Hotz, M.A.; Gong, J.; Traganos, F.; Darzynkiewicz, Z. Flow Cytometric Detection of Apoptosis: Comparison of the Assays of in Situ DNA Degradation and Chromatin Changes. Cytometry 1994, 15, 237–244. [Google Scholar] [CrossRef]
- Talevi, A.; Bellera, C.L. Challenges and Opportunities with Drug Repurposing: Finding Strategies to Find Alternative Uses of Therapeutics. Expert Opin. Drug Discov. 2020, 15, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Drug Repurposing Market-Global Market Share, Trends, Analysis and Forecast, 2023–2032. Available online: https://www.insightslice.com/drug-repurposing-market#:~:text=The%20global%20drug%20repurposing%20market,therapeutical%20uses%20for%20existing%20drugs (accessed on 7 February 2023).
- Shakil, S.; Baig, M.H.; Tabrez, S.; Rizvi, S.M.D.; Zaidi, S.K.; Ashraf, G.M.; Ansari, S.A.; Khan, A.A.P.; Al-Qahtani, M.H.; Abuzenadah, A.M.; et al. Molecular and Enzoinformatics Perspectives of Targeting Polo-like Kinase 1 in Cancer Therapy. Semin. Cancer Biol. 2019, 56, 47–55. [Google Scholar] [CrossRef]
- Cholewa, B.D.; Liu, X.; Ahmad, N. The Role of Polo-like Kinase 1 in Carcinogenesis: Cause or Consequence? Cancer Res. 2013, 73, 6848–6855. [Google Scholar] [CrossRef] [Green Version]
- Chiappa, M.; Petrella, S.; Damia, G.; Broggini, M.; Guffanti, F.; Ricci, F. Present and Future Perspective on PLK1 Inhibition in Cancer Treatment. Front. Oncol. 2022, 12, 903016. [Google Scholar] [CrossRef]
- Su, S.; Chhabra, G.; Singh, C.K.; Ndiaye, M.A.; Ahmad, N. PLK1 Inhibition-Based Combination Therapies for Cancer Management. Transl. Oncol. 2022, 16, 101332. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, R.E.A.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kothe, M.; Kohls, D.; Low, S.; Coli, R.; Cheng, A.C.; Jacques, S.L.; Johnson, T.L.; Lewis, C.; Loh, C.; Nonomiya, J.; et al. Structure of the Catalytic Domain of Human Polo-like Kinase 1’. Biochemistry 2007, 46, 5960–5971. [Google Scholar] [CrossRef]
- Liang, H.; Liu, H.; Kuang, Y.; Chen, L.; Ye, M.; Lai, L. Discovery of Targeted Covalent Natural Products against PLK1 by Herb-Based Screening. J. Chem. Inf. Model. 2020, 60, 4350–4358. [Google Scholar] [CrossRef] [PubMed]
- Keikha, M.; Esfahani, B. The Relationship between Tuberculosis and Lung Cancer. Adv. Biomed. Res. 2018, 7, 58. [Google Scholar] [CrossRef]
- Cabrera-Sanchez, J.; Cuba, V.; Vega, V.; van der Stuyft, P.; Otero, L. Lung Cancer Occurrence after an Episode of Tuberculosis: A Systematic Review and Meta-Analysis. Eur. Respir. Rev. 2022, 31, 220025. [Google Scholar] [CrossRef]
- Qin, Y.; Chen, Y.; Chen, J.; Xu, K.; Xu, F.; Shi, J. The Relationship between Previous Pulmonary Tuberculosis and Risk of Lung Cancer in the Future. Infect. Agent. Cancer 2022, 17, 20. [Google Scholar] [CrossRef]
- Fujita, M.; Oshima, T.; Morimoto, H. Benzohydrazide Derivative for Inducing g2/m Phase Arrest and Cell Death. Patent No WO2013061669A1, 2 May 2013. [Google Scholar]
- Wang, G.; He, M.; Liu, W.; Fan, M.; Li, Y.; Peng, Z. Design, synthesis and biological evaluation of novel 2-phenyl-4,5,6,7-tetrahydro-1H- indole derivatives as potential anticancer agents and tubulin polymerization inhibitors. Arab. J. Chem. 2022, 15, 103504. [Google Scholar] [CrossRef]
- Parrino, B.; Ullo, S.; Attanzio, A.; Spanò, V.; Cascioferro, S.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Cirrincione, G.; Diana, P. New Tripentone Analogs with Antiproliferative Activity. Molecules 2017, 22, 2005. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, R.; Ni, H. Eriodictyol Exerts Potent Anticancer Activity against A549 Human Lung Cancer Cell Line by Inducing Mitochondrial-Mediated Apoptosis, G2/M Cell Cycle Arrest Vorinostat enhances the therapeutic potential of Erlotinib via MAPK in lung cancer cells and Inhibition of m-TOR/PI3K/Akt Signalling Pathway. Arch. Med. Sci. 2019, 16, 446–452. [Google Scholar] [CrossRef]
- Song, Z.; Yin, Y.; Hao, S.; Wei, J.; Liu, B.; Huang, X.; Gao, C.; Zhu, R.; Liao, W.; Cai, D. JS-K induces G2/M phase cell cycle arrest and apoptosis in A549 and H460 cells via the p53/p21WAF1/CIP1 and p27KIP1 pathways. Oncol. Rep. 2019, 41, 3475–3487. [Google Scholar] [CrossRef]
- Alqosaibi, A.I.; Abdel-Ghany, S.; Al-Mulhim, F.; Sabit, H. Vorinostat Enhances the Therapeutic Potential of Erlotinib via MAPK in Lung Cancer Cells. Cancer Treat. Res. Commun. 2022, 30, 100509. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, a potential target for cancer therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmit, T.L.; Zhong, W.; Setaluri, V.; Spiegelman, V.S.; Ahmad, N. Targeted depletion of Polo-like kinase (Plk) 1 through lentiviral shRNA or a small-molecule inhibitor causes mitotic catastrophe and induction of apoptosis in human melanoma cells. J. Investig. Dermatol. 2009, 129, 2843–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.; Kraikivski, P.; Shafiekhani, S.; Terhune, S.S.; Dash, R.K. Crosstalk between Plk1, p53, cell cycle, and G2/M DNA damage checkpoint regulation in cancer: Computational modeling and analysis. Npj Syst. Biol. Appl. 2021, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Pezuk, J.A.; Brassesco, M.S.; Morales, A.G.; de Oliveira, J.C.; de Oliveira, H.F.; Scrideli, C.A.; Tone, L.G. Inhibition of polo-like kinase 1 induces cell cycle arrest and sensitizes glioblastoma cells to ionizing radiation. Cancer Biother. Radiopharm. 2013, 28, 516–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Yu, H.; Li, W.; Li, F.; Wang, S.; Yu, N.; Jiang, Q. Plk1 promotes the migration of human lung adenocarcinoma epithelial cells via STAT3 signaling. Oncol. Lett. 2018, 16, 6801–6807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunasekaran, P.; Lee, G.H.; Hwang, Y.S.; Koo, B.C.; Han, E.H.; Bang, G.; La, Y.K.; Park, S.; Kim, H.N.; Kim, M.H.; et al. An investigation of Plk1 PBD inhibitor KBJK557 as a tumor growth suppressor in non-small cell lung cancer. J. Anal. Sci. Technol. 2022, 13, 1–10. [Google Scholar] [CrossRef]
- Zhang, R.; Xu, Z.; Ning, F.; Kong, Y.; Li, X.; Wang, X.; Yu, X. The effect of inhibition of Plk1 expression on the sensitivity and metastasis of cisplatin in lung cancer cell line A549. Acta Med. Mediterr. 2020, 36, 2833–2838. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Code | Name of Compound | Binding Affinity |
|---|---|---|
| C1 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-2,4-dichlorobenzohydrazide | −9 kcal/mol |
| C2 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-2-aminobenzohydrazide | −8.8 kcal/mol |
| C3 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-2-bromobenzohydrazide | −8.9 kcal/mol |
| C4 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-2-chlorobenzohydrazide | −9 kcal/mol |
| C5 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-2-methylbenzohydrazide | −8.9 kcal/mol |
| C6 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-3-acetylbenzohydrazide | −9 kcal/mol |
| C7 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-3-aminobenzohydrazide | −7.8 kcal/mol |
| C8 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-3-chlorobenzohydrazide | −9.7 kcal/mol |
| C9 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-3-methylbenzohydrazide | −9.1 kcal/mol |
| C10 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-3-nitrobenzohydrazide | −9 kcal/mol |
| C11 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-4-aminobenzohydrazide | −8.8 kcal/mol |
| C12 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-4-bromobenzohydrazide | −8.9 kcal/mol |
| C13 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-4-chlorobenzohydrazide | −9 kcal/mol |
| C14 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-4-fluorobenzohydrazide | −8.8 kcal/mol |
| C15 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-4-hydroxybenzohydrazide | −8 kcal/mol |
| C16 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-4-methoxybenzohydrazide | −8.9 kcal/mol |
| C17 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-4-methylbenzohydrazide | −8.7 kcal/mol |
| C18 | N′-(4-(1H-pyrrol-1-yl)benzoyl)-4-nitrobenzohydrazide | −9.6 kcal/mol |
| C19 | N′-benzoyl-4-(1H-pyrrol-1-yl)benzohydrazide | −9 kcal/mol |
|
Control (BI2536) | 4-{[(7R)-8-cyclopentyl-7-ethyl-5-methyl-6-oxo-5,6,7,8-tetrahydropteridin-2-yl]amino}-3-methoxy-N-(1-methylpiperidin-4-yl)benzamide | −9.3 kcal/mol |
| Native Ligand | 4-(4-methylpiperazin-1-yl)-N-{5-[2-(thiophen-2-yl)acetyl]-1H,5H-pyrrolo[3,4-c]pyrazol-3-yl}benzamide | −8.6 kcal/mol |
| Ligands | Binding Energy | Interacting Amino Acids of PLK1 |
|---|---|---|
| C8 | −9.7 kcal/mol | Leu59, Cys67, Ala80, Val114, Leu130, Glu131, Leu132, Cys133 *, Arg134, Arg136, Phe183, Gly193, Asp194 |
| C18 | −9.6 kcal/mol | Leu59, Cys67, Ala80, Val114, Leu130, Glu131, Leu132, Cys133 *, Arg134, Arg136, Phe183, Gly193, Asp194 |
| Positive control (BI2536) | −9.3 kcal/mol | Gly60, Lys61, Gly62, Gly63, Phe64, Lys82 *, Val84, Lys97, Phe183, Asp194 |
| Redocked Native ligand (DB07186) | −8.6 kcal/mol | Lys61, Gly62, Gly63, Phe64, Cys67, Lys82, Val114, Glu131, Cys133, Arg136, Phe183, Asp194, Gly 196 |
| Compounds | IC50 (µM) | ||
|---|---|---|---|
| MCF7 | A549 | HepG2 | |
| C8 | 10.51 ± 1.9 | 9.54 ± 1.1 | 10.82 ± 1.3 |
| C18 | 12.34 ± 0.9 | 10.38 ± 1.7 | 11.41 ± 2.5 |
| Doxorubicin * | 10.96 ± 1.6 | 8.20 ± 0.9 | 9.21 ± 1.0 |
| Cell Cycle Stage | Untreated | C8 (10 µM) |
|---|---|---|
| Sub G0/G1 | 0.53 | 1.63 |
| G0/G1 | 88.79 | 31.22 |
| S | 1.03 | 12.44 |
| G2/M | 9.34 | 39.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hagbani, T.A.; Moin, A.; Hussain, T.; Gupta, N.V.; Alshammari, F.; Rizvi, S.M.D.; Dixit, S. Anticancer Activity of Anti-Tubercular Compound(s) Designed on Pyrrolyl Benzohydrazine Scaffolds: A Repurposing Study. Processes 2023, 11, 1889. https://doi.org/10.3390/pr11071889
Hagbani TA, Moin A, Hussain T, Gupta NV, Alshammari F, Rizvi SMD, Dixit S. Anticancer Activity of Anti-Tubercular Compound(s) Designed on Pyrrolyl Benzohydrazine Scaffolds: A Repurposing Study. Processes. 2023; 11(7):1889. https://doi.org/10.3390/pr11071889
Chicago/Turabian StyleHagbani, Turki Al, Afrasim Moin, Talib Hussain, N. Vishal Gupta, Farhan Alshammari, Syed Mohd Danish Rizvi, and Sheshagiri Dixit. 2023. "Anticancer Activity of Anti-Tubercular Compound(s) Designed on Pyrrolyl Benzohydrazine Scaffolds: A Repurposing Study" Processes 11, no. 7: 1889. https://doi.org/10.3390/pr11071889
APA StyleHagbani, T. A., Moin, A., Hussain, T., Gupta, N. V., Alshammari, F., Rizvi, S. M. D., & Dixit, S. (2023). Anticancer Activity of Anti-Tubercular Compound(s) Designed on Pyrrolyl Benzohydrazine Scaffolds: A Repurposing Study. Processes, 11(7), 1889. https://doi.org/10.3390/pr11071889