1. Introduction
Many substances used or produced in industries pose a challenge in terms of disposal and impact on the environment due to their flammability, toxicity or volatility. Environmentally friendly processes in the chemical industry are one way to reduce emissions to the atmosphere and contamination of wastewater. Many organic solvents used in the chemical industry are harmful to health, carcinogenic or environmentally hazardous, which makes the importance of developing environmentally friendly processes even more important. The use of non-toxic solvents in the extraction and recovery of valuable substances offers a crucial opportunity here [
1]. Lignin and extractives are by-products of the pulping process. The material use of lignin as a component of cardboard, resins or fillers has been established for a long time. The combination of lignin and extractives for use in the pharmaceutical and cosmetics industry is an innovation in this context. Bio-based phenolic compounds found in spruce bark are known to be value-added products with antioxidant capacity. The gain and isolation of these extractive compounds require suitable separation techniques. In recent decades, research into lignocellulosic biorefinery has focused on the contained aromatic compounds of agricultural and forestry waste [
2]. In this context, the valorisation of process waste streams is a central objective. The organosolv process was chosen for this study because it improves the recovery of various phenolic and flavonoid substances. The advantage is that good yields of desired aromatic compounds are already achieved at low temperatures between 50 and 95 °C. This is particularly relevant in terms of energy and costs [
3]. Bark, in particular, is currently regarded as one of the most abundant raw materials, since spruce and pine are usually used in the pulp, paper and woodworking industries. Greenwood of conifers contains around 10% bark, and per year, there is around 500,000 m
3 of spruce bark (density = 380 kg m
−3) in Slovakia and 900,000 t of bark per year in the Finnish forest industry [
4]. In Germany, around 11% of the tree volume of conifers is determined as material loss during debarking of the wood logs [
5]. In Central and Northern Europe, around 25 Mio m
3 of bark of softwood conifer trees are available [
6]. At present, most bark waste is combusted for heat or electricity, although it contains various useful aromatic compounds such as lignans, tannins and stilbenes [
7]. Jablonsky et al. provided a comprehensive review of the extractive properties of softwood bark and of its valorisation by extraction of utilisable chemicals [
7]. The extraction of phenolic components is also in line with Goal 8, “Sustainable Economic Growth”, and Goal 15, “Promote sustainable use of terrestrial ecosystems” of the SDG 2030 agenda. In this context, the substitution of fossil chemicals with bio-based chemicals is one of the most important tasks for the future [
8].
Extraction and purification of these desired compounds are the key tasks in the side-stream valorisation of forestry waste. Organosolv solvents are the preferred process media for biorefining lignocellulosic feedstocks (e.g., wood) [
9] and thus for bark, and were, therefore, the solvents of choice when working with tree bark. These bioactive compounds also exhibit anti-bacterial, anti-inflammatory, anti-viral, anti-malarial, anti-mutagenic, anti-tumour, cytotoxic, fungicidal, insecticidal, and pharmacokinetic activities and properties. The extraction of hydrophobic structures, such as phenolics and flavonoids, is of particular interest because their antioxidant capacity promises a wide range of potential applications [
10]. There are various studies comparing different extraction techniques, such as pressurised liquid extraction (PLE), using different solvents, supercritical fluid extraction (SFE), microwave-assisted extraction (MAE) or reflux boiling (RB) [
11]. Hot-water extraction processes, either under or without pressure, are well-known methods for isolating tannins from bark [
12].
Co et al. showed that the yield of bark extraction is increased by using ethanol as a solvent which was one of the reasons for using this solvent in our present study [
13]. Krogell et al. and Kreps et al. reported a method that uses hexane and acetone to extract aromatic compounds from spruce bark [
7,
8]. Burčova et al. used both ethanol and n-hexane as solvents in their Soxhlet bark extractions [
14]. Jablonsky successfully obtained higher yields of phenolic compounds out of spruce bark by the use of deep eutectic solvents instead of conventional solvents [
15]. Spinelli shows in her works that ultrasonic-assisted extraction shows a higher yield of transresveratrol compared to established pressurised liquid extraction [
16]. All these studies were performed using accelerated extraction methods in laboratory experiments, e.g., ultrasonic or microwave assistance. The goal of our study was to implement an easier experimental setup that can be easily adapted by industrial partners. Throughout all the studies, we intended to use ethanol/water for solvent mixtures in our experiments due to their environmentally friendly and non-toxic properties.
The phenolic compounds are highly protective against bacteria and fungi and can act as antiproliferative compounds and as antioxidants, and exhibit 2,2-diphenyl-1-picrylhydrazyl (DPPH) free-radical scavenging activity [
17,
18]. Aufischer et al. found that phenolic hydroxyl groups in lignin and its degradation products react with free radicals and trap them within the sterically hindered phenolic structure [
19]. Using the DPPH method, they compared lignin and its degradation products with the commercial reference substances butylated hydroxytoluene (BHT), butylated hydroxyanisole (BHA), and Irganox 1010 in terms of antioxidant activity [
19]. The antioxidant activities of the degraded lignin fractions were better than those of BHT and BHA, and on par with commercially available Irganox 1010 [
19]. Alzagameem et al. used various assays to examine the extracts from spruce bark waste for antioxidant activity of bioactive constituents and found correlations between antioxidant activities and minor structural differences of purified lignins [
20]. The biomass source, the pulping process and the degree of purification have an influence on antioxidant activity [
20]. Strižincová presented in her studies that the antioxidant activity not only depends on the concentration of phenolic compounds in the extract, but also on the sample composition [
21].
Due to the better environmental impact and overall economical extract utilisation, this study focused on a systematic view of the extraction process of spruce bark and demonstrated the impacts of temperature and solvent polarity using various ethanol/water ratios. Our results form the basis for pilot plant and industrial applications. We used mild conditions and a relatively simple processing setup to address aspects of economical and cost-efficient upscaling. As part of designing an improved biorefinery, we investigated the total phenolic content as a function of solvent polarity, temperature, and time. Further, the antioxidant activity of the extracts alone and in combination with degraded lignin was studied.
3. Materials and Methods
3.1. Materials and Reagents
Spruce bark was purchased from SAPPI (Gratkorn, Austria). After drying at 90 °C, the bark was milled with a cutting mill and separated using a 1 mm sieve. Ethanol, anhydrous pyridine, aluminium chloride, sodium acetate, vanillic acid and p-hydroxybenzoic acid were obtained from Carl Roth (Karlsruhe, Germany). Bis(trimethylsilyl)-trifluoroacetamide (BSTFA), Folin-Ciocalteu reagent, ferulic acid, p-coumaric acid, cinnamic acid, o-hydroxybenzoic acid, quercetin and gallic acid were purchased from Sigma (Taufkirchen, Germany). Glucose, mannose, galactose, arabinose, and xylose were acquired from Alfa Aesar (Karlsruhe, Germany).
3.2. Extraction Method
Organosolv extraction was performed in a PARR 4560 autoclave with a volume of 450 mL (PARR Germany Frankfurt/Main, Germany). The set-up enabled monitoring of time, temperature, pressure, heating, and stirring velocity. Extractions were performed with a solid/liquid ratio of 1:20. In each case, 5 g of spruce bark was examined with specified solvent mixtures of ethanol/water: 0% (v/v), 25% (v/v), 50% (v/v) and 75% (v/v). The stirring was kept at 400 rpm. The extraction time was 2 h for all experiments, and the temperature was set to 40, 60, 80 and 100 °C. All extraction conditions were set to mimic ethanol-water pretreatments of bark in the biorefinery industry with Organosolv media.
Each experiment was conducted three times. After extraction, the crude suspension was filtered through a funnel with porosity 3. Residual bark was dried at 90 °C for 1.5 h, and analysed gravimetrically.
3.3. Determination of Extractives Yield
The yield of extractives (YE, %) in each experiment was determined by drying the bark samples at 105 °C to a constant weight. The results are expressed according to Strizincova et al. based on the dry matter weighed before and after extraction [
21]:
where m
i is the dry mass (g) of the bark before extraction and m
j is the mass (g) of the bark after extraction and drying.
3.4. Determination of Chemical Composition
Ash content, acid-insoluble lignin, acid-soluble lignin and saccharides were determined according to NREL/TP-510-42618 [
22,
23,
24]. In total, 300 mg of milled bark was suspended in 3 mL of 72 wt.% sulphuric acid at 30 °C in a heater bath. After a storage time of 1 h, 84 mL of distilled water was added to the sample tubes, which were heated to and then kept at 121 °C in an autoclave for 1 h. After cooling down, the suspension was filtered using porous crucibles. The acid-soluble lignin content was determined based on the absorbance at 215 nm wavelength. The acid-insoluble lignin (Klason lignin) content was determined gravimetrically after drying the crucibles at 105 °C (24 h). Carbohydrate monomers were quantified by High-Performance Anion Exchange Chromatography (HPAEC).
3.5. Determination of Total Phenolics Content (TPC)
1 mL of extract and 5 mL of Folin-Ciocalteu reagent were added to a 25 mL flask and dissolved with 0.1 M sodium hydroxide. In total, 10 mL of distilled water was added, and the samples were stored for 5 min for dissolution. In total, 5 mL of 20 wt.% sodium carbonate (Na
2CO
3) solution was added to the 50 mL flask containing the sample solution, which was then filled up with distilled water. Sample solutions were then stored for 2 h at room temperature and mixed every 20 min. The total phenolic content was determined based on the absorbance at 760 nm wavelength in polystyrene cuvettes, as described by Talmaciu et al. [
32]. All UV/VIS measurements were performed twice on a Thermo Scientific Multiscan Go Spectrophotometer. In total, 10 mg of gallic acid was dissolved in a 50 mL flask of distilled water and used as a calibration substance. A 5-point calibration line of 10, 20, 40, 100 and 200 mg L
−1 was used for quantification.
3.6. Determination of Total Flavonoid Content (TFC)
The TFC was determined as described in the literature [
32]. In total, 5 mL of extracts and 15 mL of 96 wt.% ethanol were added into a 50 mL flask. After 10 min of dissolution time, 1 mL of aluminium(III) chloride-ethanol with a ratio of 10 wt.% (
w/
v) and 1 mL of 0.1 M sodium acetate solution was added to the sample. The flask was filled up with distilled water. The sample solution was stored for 2 h at room temperature. The total flavonoid content was determined in two parallel measurements based on the absorbance at 415 nm wavelength in polystyrene cuvettes. In total, 5 mg quercetin in a 50 mL flask was used as the standard solution at different dilutions (10, 25, 50, 75, 100 mg L
−1) to produce a calibration curve.
3.7. Determination of Total Tannin Content (TTC)
To 1 mL of extract, 0.5 mL of Folin-Ciocalteu reagent was added in a 25 mL flask. In total, 1 mL of 20 wt.% Na
2CO
3 solution was added, and the flask was filled up with distilled water. After incubation for 2 h, the absorbance of the solution at 760 nm was measured three times. The TTC was determined using a calibration curve obtained with tannic acid (TA) in standard concentrations of 10, 20, 50, 100, and 200 mg L
−1. Results were expressed in milligrams of tannic acid per gram of dry bark in accordance with the literature [
32].
3.8. Determination of Saccharide’s Content
All liquid samples were diluted 1:100, and the saccharides contained were quantified by HPAEC with pulsed amperometric detection (HPAEC-PAD). All measurements were performed on a Dionex 5000+ (ThermoFisher Scientific, Waltham, MA) equipped with a CarboPac20 column (particle size = 6 µm, pore size ≤ 10 Å, crosslinking = 55%, ion exchange capacity = 65 µeq) and a VWD detector. A total of 1.5 mM NaOH was used as solvent A and 200 mM NaOH was used as solvent B. The flow rate was set isocratically with 0.8 mL min−1 of solvent ratio A/B = 80:20. Saccharides were quantified by a 5-point calibration. Each sample was measured three times, and the standard deviation was below 5%.
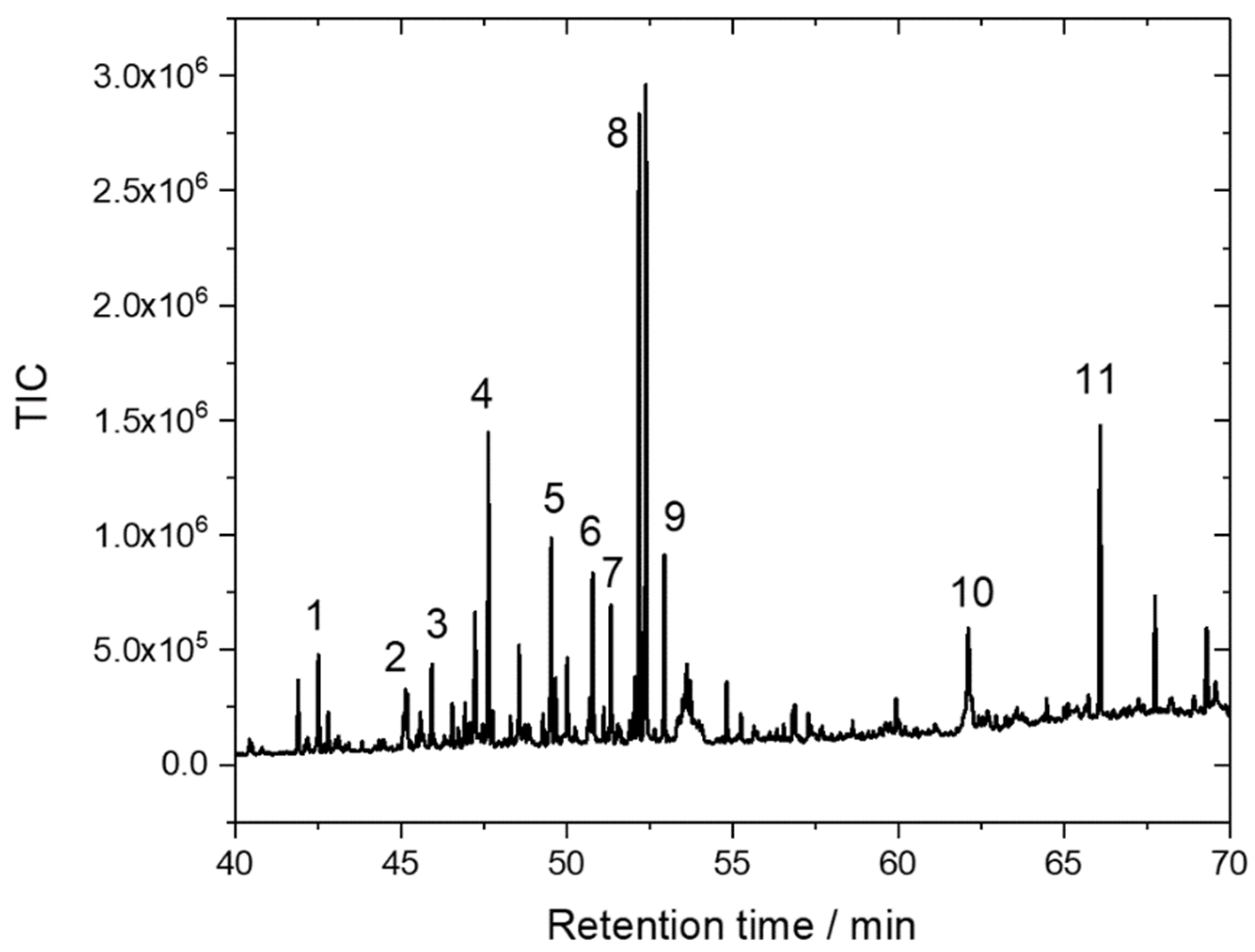
3.9. GC-MS Determination of Phenolic Compounds
200 µL of extract was pipetted into a 1.5 mL GC vial and subjected to lyophilisation at −54 °C for 48 h. In total, 900 µL of anhydrous pyridine was added to the sample and kept under 70 °C for 1 h. The sample was cooled down to room temperature, 100 µL BSTFA was added for derivatisation, and the mixture was heated to 70 °C for 1 h. After cooling down, samples were separated by gas chromatography and identified by mass spectrometry. GC-MS measurements were conducted on a Shimadzu QP2010 gas chromatograph coupled with a Shimadzu QP2020 Dual Stage Mass Spectrometer. An HP5-MS column (60 m length × 0.25 mm inner diameter × 0.25 µm film thickness; J&W Scientific, Folsom, CA, USA) was used. The injection port was operated under the following conditions: split injection, constant column flow: 1.2 mL min
−1 with helium carrier gas, purge flow: 3.0 mL min
−1, split ratio: 1:10, total flow: 15.2 mL min
−1; injector temperature: 250 °C constant. Column temperature gradient profile: 50 °C (15 min), followed by 5 °C min
−1 to 300 °C (5 min). The mass-spectrometric detector was operated in EI mode (70 eV ionisation energy at 1.13 × 10
−7 Pa). Ion source temperature: 200 °C, transfer line: 250 °C. Data was acquired in Scan mode ranging from 45 to 500
m/
z. A total of 1 µL aliquots were injected by an AOC 6000 autosampler. The NIST/Wiley 2014 database was used for compound identification. The method was described in detail by Liftinger [
42].
3.10. DPPH Radical Scavenging Activity
The DPPH radical scavenging activity was determined according to the literature [
19]. For the DPPH stock solution, 50 mg of DPPH was dissolved in absolute ethanol. Crude bark extracts were diluted in dimethyl sulfoxide (DMSO) using solutions of 2.0 g L
−1, 1.0 g L
−1, 0.5 g L
−1, 0.25 g L
−1 and 0.125 g L
−1. In total, 100 µL of the samples and 3900 µL of the DPPH stock solution were added to 10 mL screw-cap vials. The vials were closed and stored for 90 min incubation time at room temperature. Absorption of the samples was determined at 517 nm wavelength. All measurements were performed three times. AA, AAI, and IC
50 were calculated according to Aufischer et al. [
19].
The antioxidant activity index (AAI) was calculated according to
where IC
50 is the half-maximal inhibitory concentration.
3.11. Antioxidant Activity by OIT-DSC
Oxidation induction time differential scanning calorimetry (OIT-DSC) was used to characterise bark extracts, oligolignin and Irganox 1010, according to the works of Aufischer. A stock solution of each substance was prepared by dissolving it in EtAc at a concentration of 30.0 g L−1. In total, 100 µL of each stock solution was added to 3.00 g squalane and homogenised in an ultrasonic bath. Pristine squalane and each squalane spiked with the stock solution were transferred to a Tzero pan and analysed with a DSC TA Instrument Q20. In total, 20 mg of sample material were weighed into-gold crucibles. All samples were heated from 25 °C to 190 °C using 10 °C. min−1 as heating rate. After 5 min of 190 °C, the nitrogen (flow rate = 50 mL·min−1) was switched to oxygen (50 mL·min−1). Heat and oxygen flow were constantly kept for 100 min.
3.12. Molecular Weight Distribution
All samples were analysed for their molecular weight by Size Exclusion Chromatography. A total of 1 mg lignin sample is dissolved in 0.1 M NaOH and filtered through a 0.45 µm syringe filter. Polystyrene standards are prepared the same way.
Molar mass distributions of the lignin samples were performed on a Thermo Fisher Dionex ICS 5000+. The chromatographic system consists of a pre-column PSS MCX Guard 50 × 8 mm and three analytical (PSS Analytical 100 A, 300 × 8 mm; PSS Analytical 1000 A, 300 × 8 mm; PSS Analytical 100,000 A, 300 × 8 mm) columns. The UV detector used is a VWD detector with 280 nm as the standard wavelength. Molar mass calibrations are carried out using Polystyrene standards (Mw= 1000 g/mol, Mw= 3400 g/mol, Mw= 10,000 g/mol, Mw= 80,000 g/mol, Mw= 140,000 g/mol).
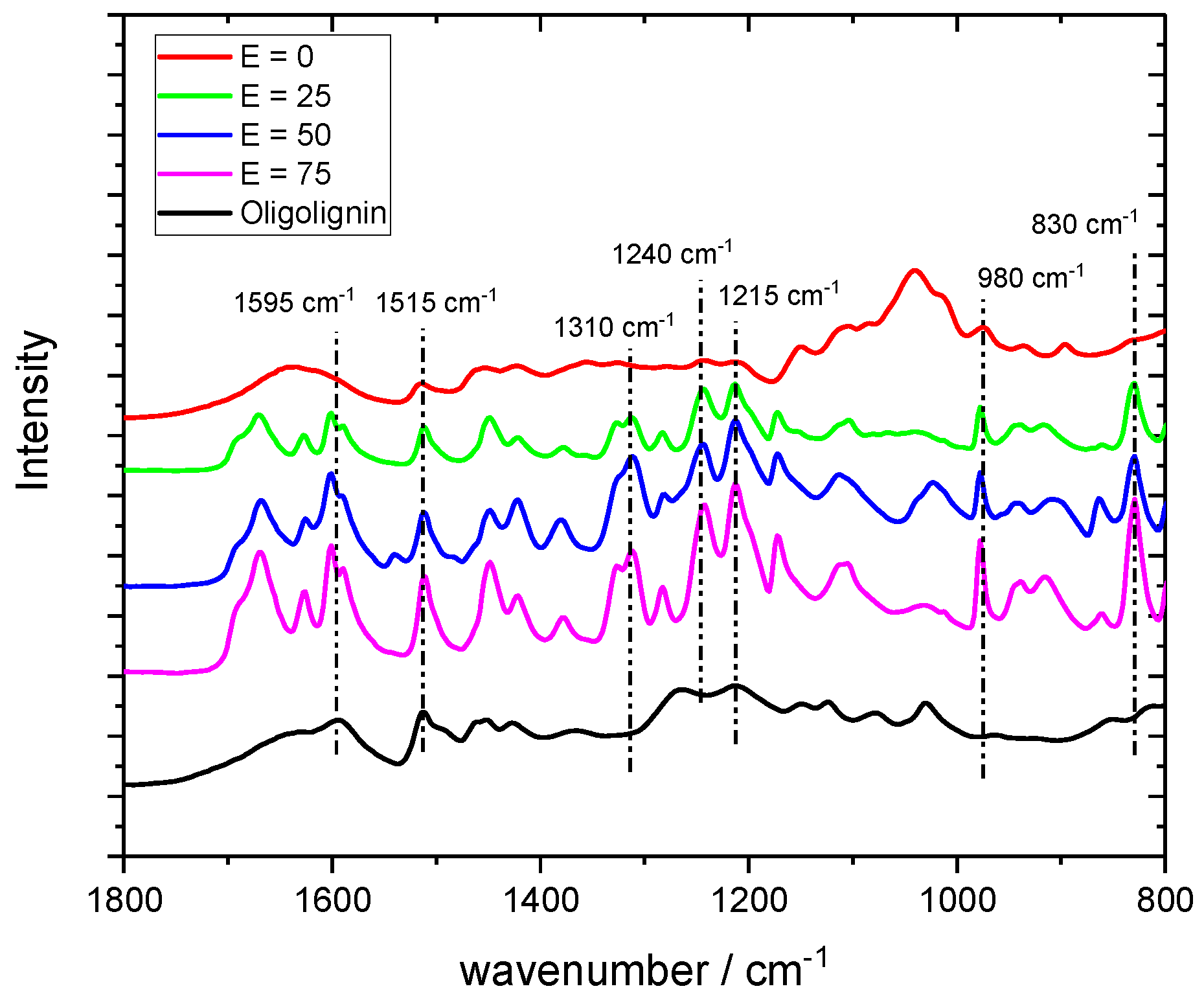
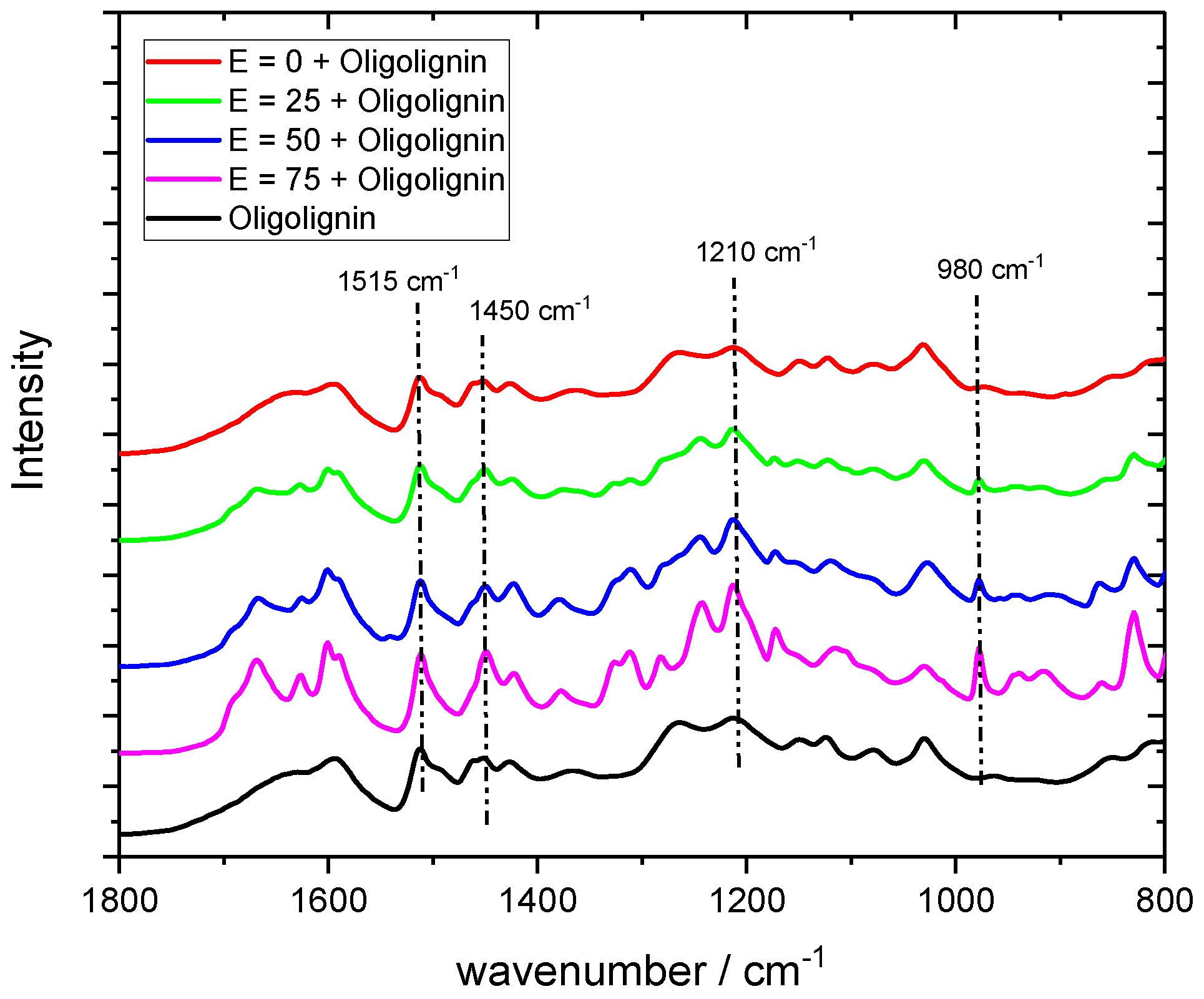
3.13. Fourier Transformation—Infrared Spectroscopy (FT-IR)
FT-IR spectra of all samples were recorded on a Bruker Vertex 70 infrared spectrometer equipped with a Pike Technologies monolithic diamond ATR. For each spectra, a resolution of 4 cm−1 and a measurement time of 32 scans with a wavenumber range of 4000 cm−1 to 600 cm−1 were used.
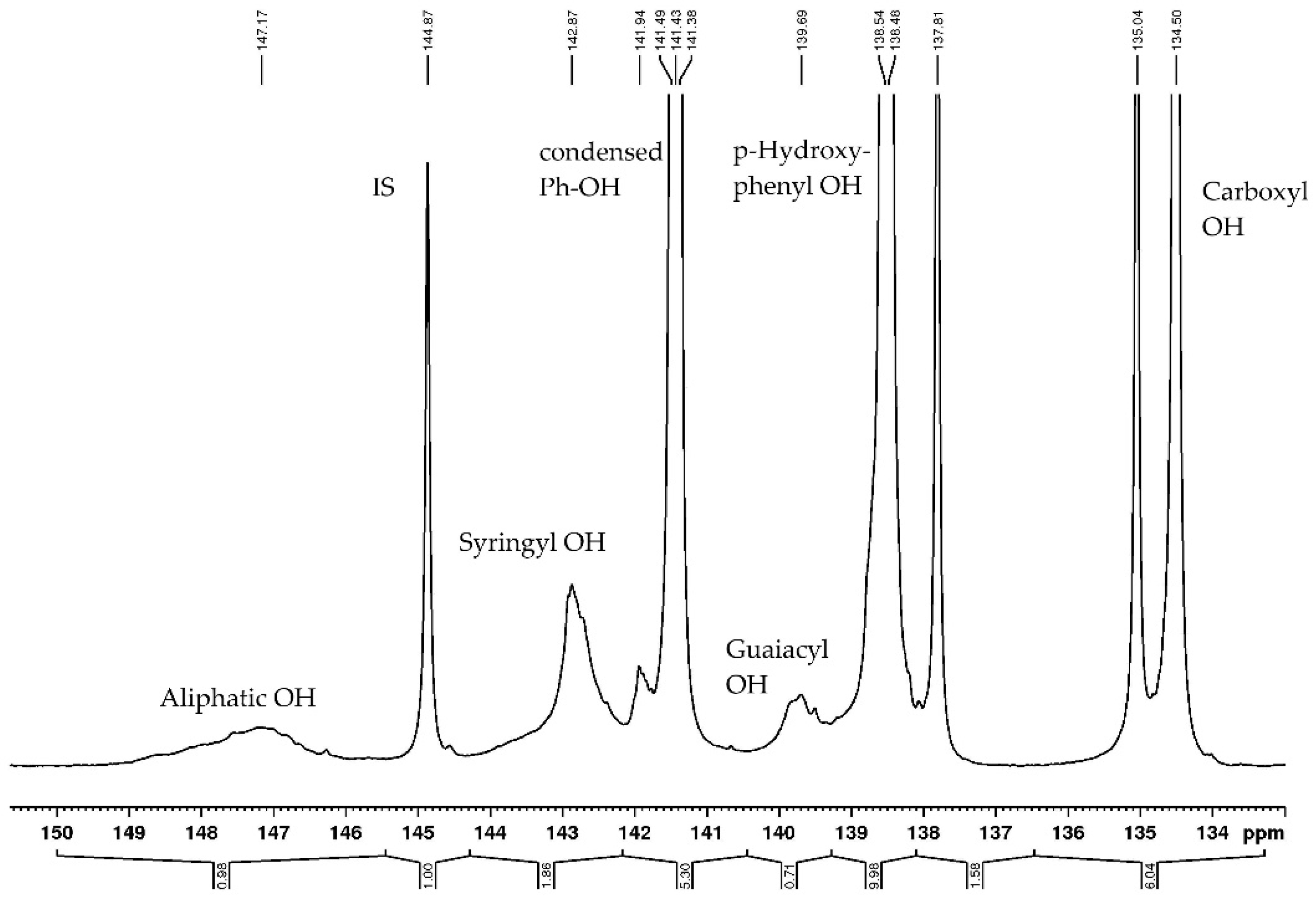
3.14. P NMR of Bark Extracts
40 mg of accurately weighted freeze-dried bark extracts were transferred into a sample vial, dissolved in 400 µL of pyridine and deuterated chloroform (1.6:1, v/v) and left at room temperature overnight with continuous stirring. Cholesterol (200 µL, 19 mg/mL) and chromium (III) acetylacetonate (50 µL, 11.4 mg/mL) were used as internal standard (IS) and relaxation reagents, respectively. After 2 h, 100 µL of phosphitylating reagent II (2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane) was added, and the mixture was transferred into a 5-mm-OD NMR tube. All measurements were performed at a Bruker 300 MHz NMR spectrometer, including the following conditions: relaxation delay = 10 s, pulsewidth = 4 us, pulseprogram = zgig (inverse gated), acquisition time = 1 s, Fid resolution = 0.5 Hz, temperature = 298 K, Offset = 153 ppm.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}