Dynamics of the Bacterial Community Associated with Phaeodactylum tricornutum Cultures

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
2.2. Growth Measurements
2.3. Genomic DNA Extraction
2.4. Barcoded 16S-V6-Next Generation Sequencing
2.5. Bioinformatics Analysis
2.6. Mathematical Model
- (1)
- (2)
- the mortality rate of each population was inversely proportional to (1 + ), to account for the fact that cells during replication (high growth rate) were healthier;
- (3)
- additional contributions to population mortality was given by the presence in the environment of noxious elements like bactericidal substances;
- (4)
- changes in metabolite concentrations are in general directly proportional to the growth of the consumers and producers;
- (5)
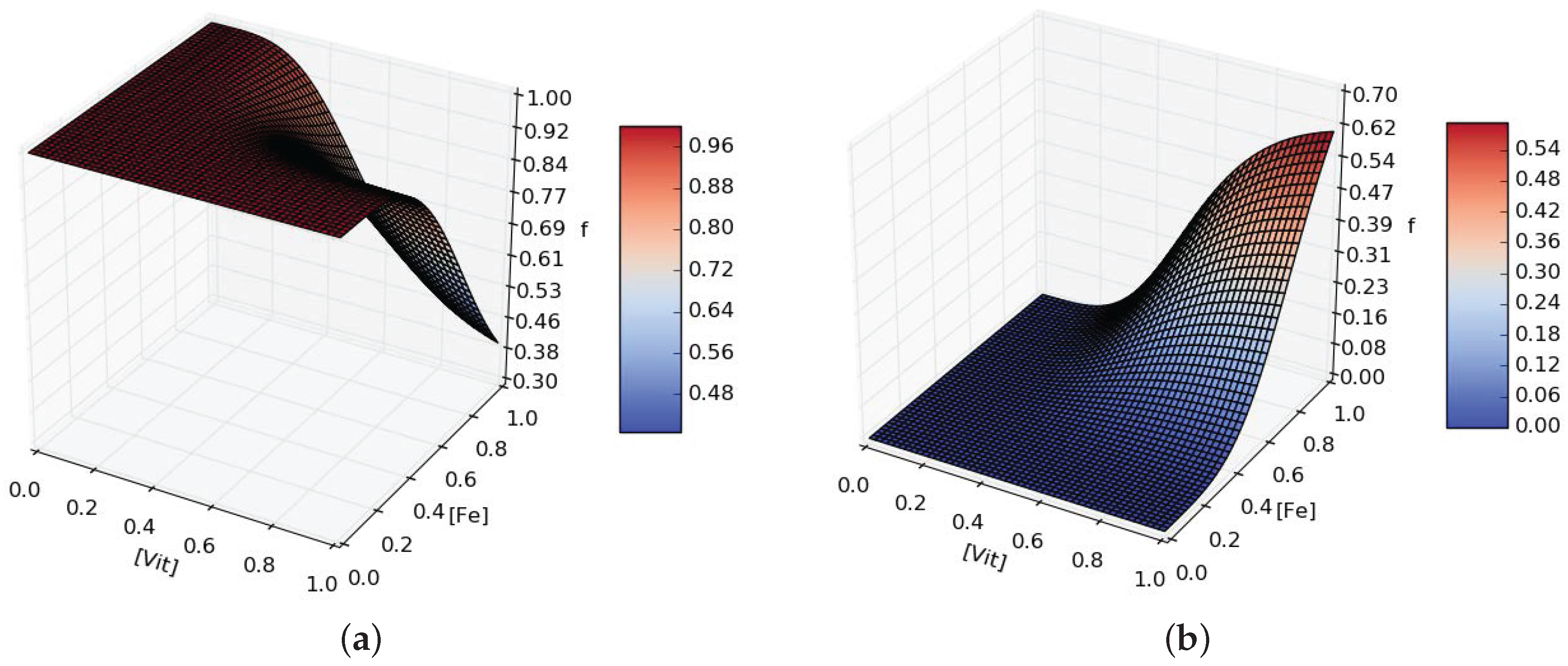
- in the event of micronutrient scarcity (Iron and Vitamins in our model), P. tricornutum will secrete more organic carbons favored by those bacteria able to provide the needed micronutrients.
- (1)
- the initial quantity of Iron and Vitamins is 10 times higher in complete media;
- (2)
- the initial quantity of P. tricornutum biomass is matched to the first data point.
3. Results
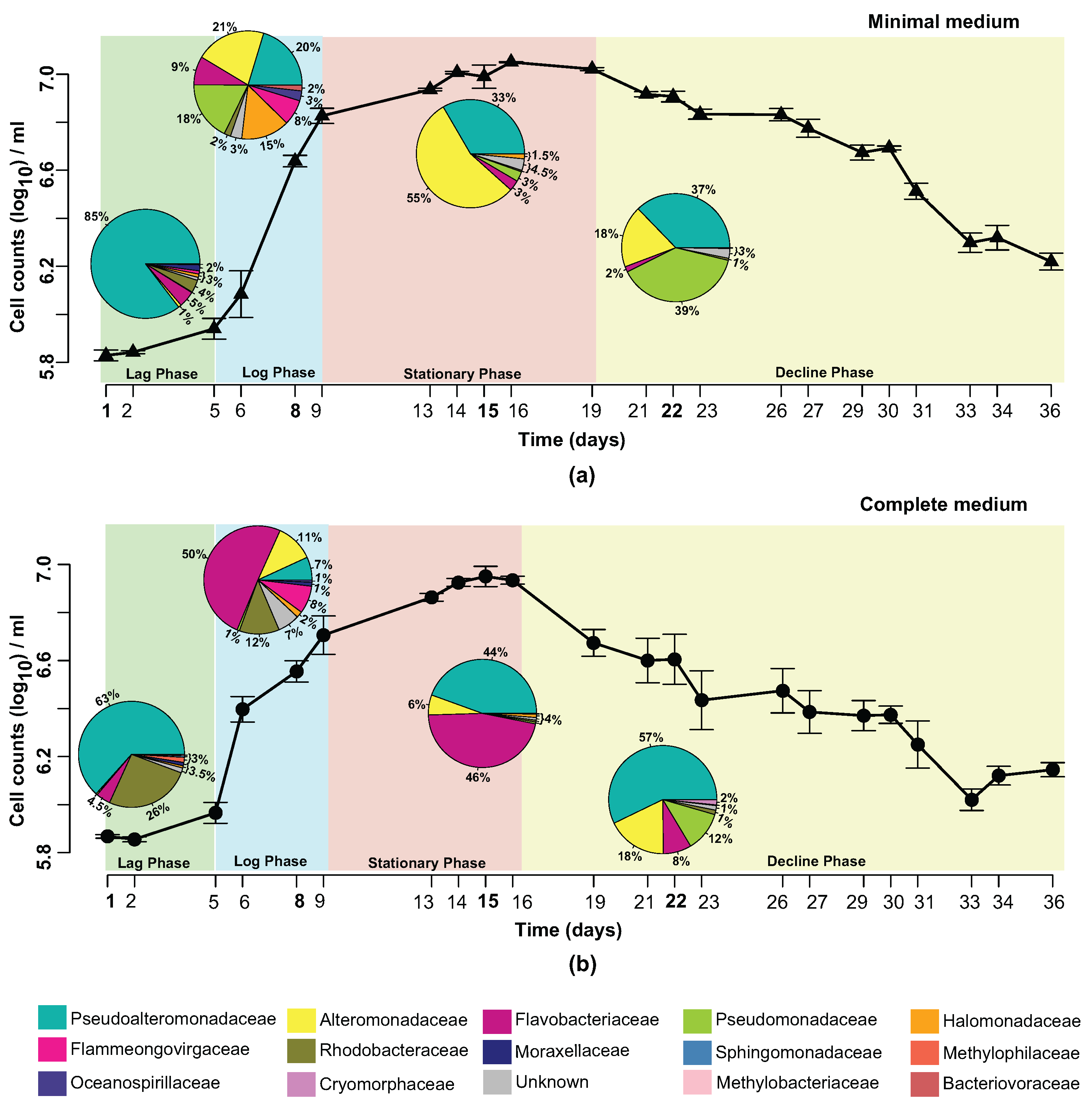
3.1. Characteristics of Phaeodactylum tricornutum Growth
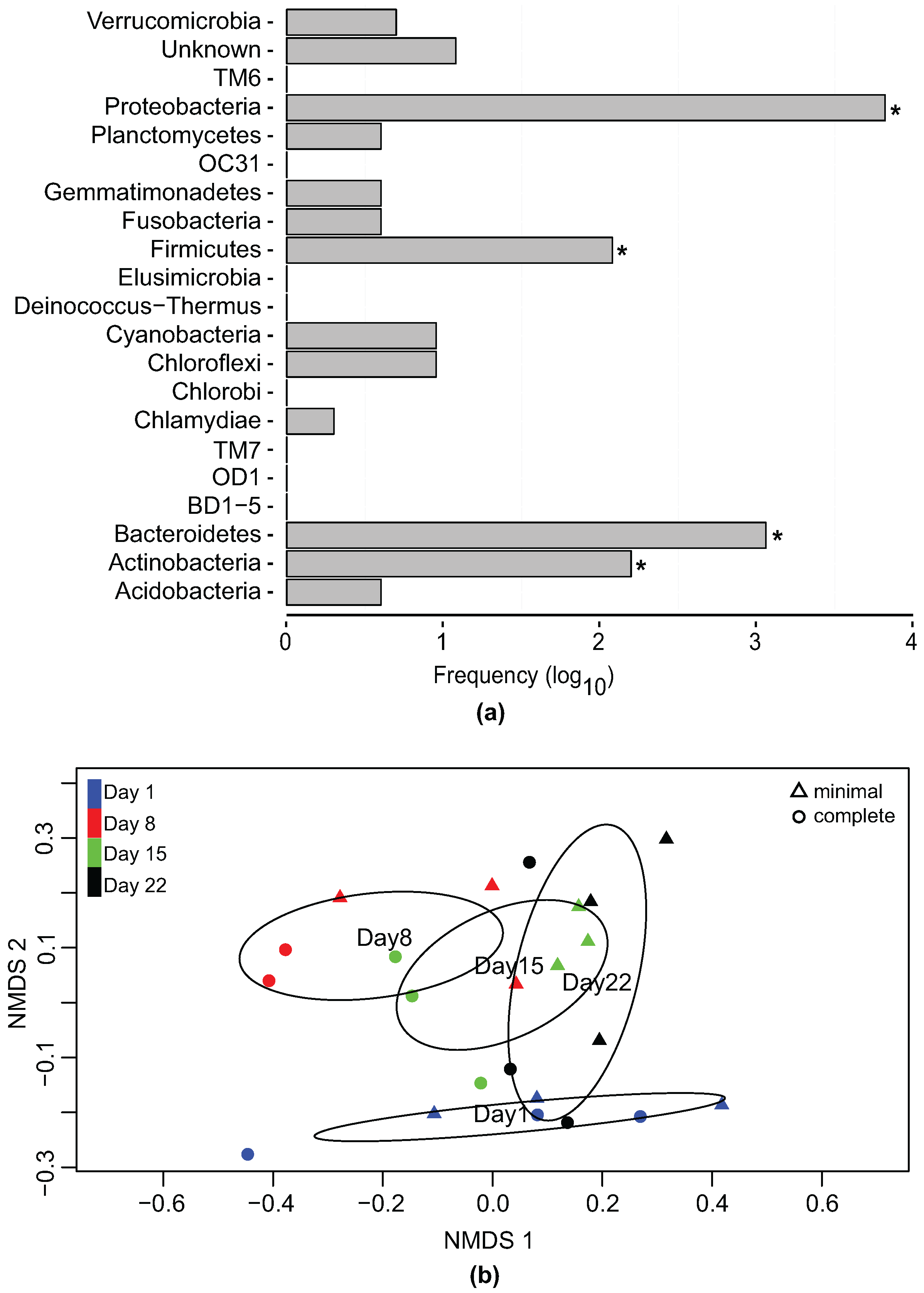
3.2. Bacterial Community Profile of Phaeodactylum tricornutum Cultures
3.3. Effect of Temporal Evolution and Media Composition on the Bacterial Community Profile
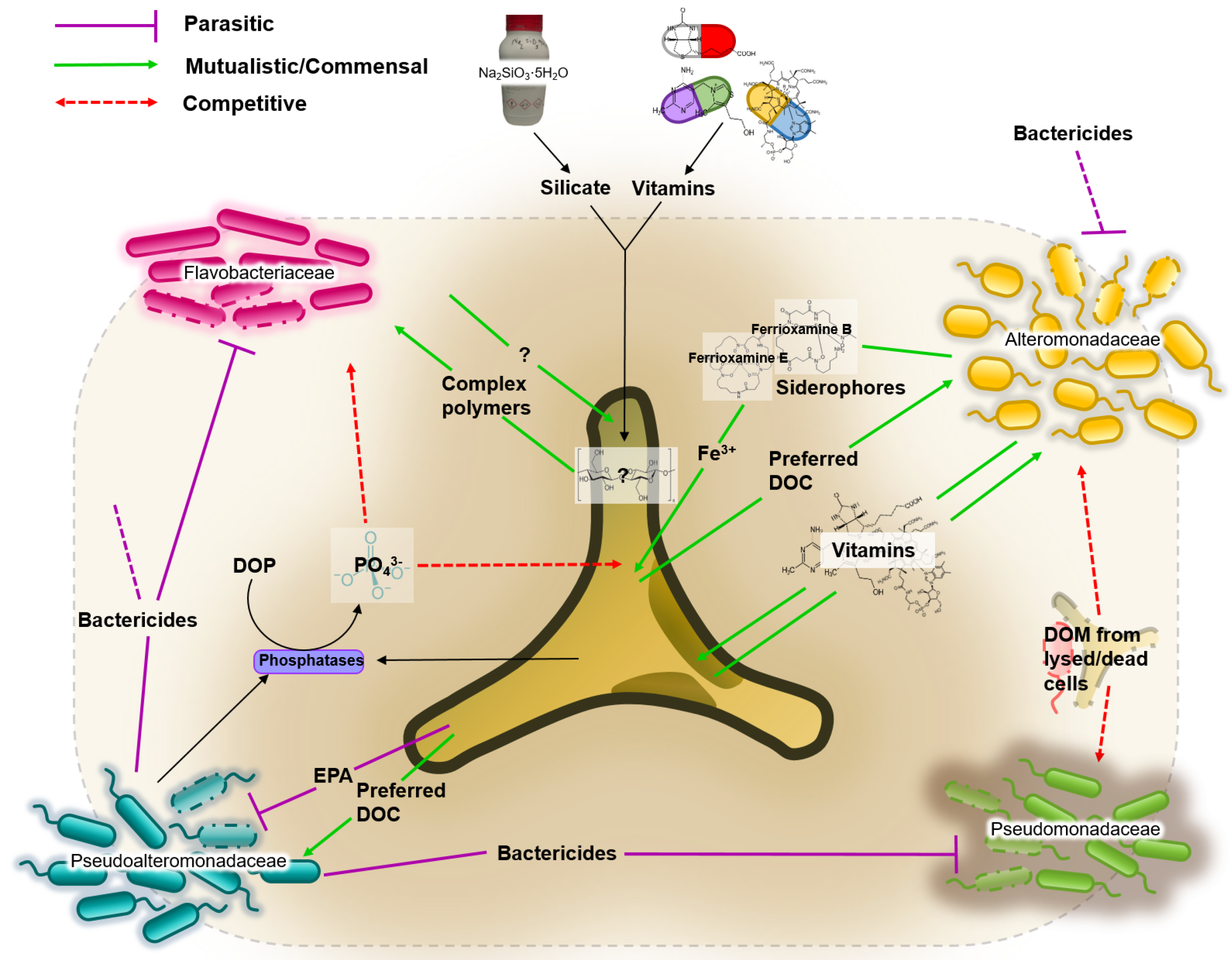
3.4. Network of Putative Interactions between Phaeodactylum tricornutum and Identified Bacterial Families
- Bactericidal metabolites. Several species of the Pseudoalteromonadaceae family have been reported to possess bactericidal effects [54]. This ability to suppress the growth of competing bacteria could explain the dominance of Pseudoalteromonadaceae in almost all cultures irrespective of media composition. P. tricornutum also demonstrates bactericidal properties by excreting fatty acids (such as eicosapentaenoic acid or EPA), nucleotides, peptides, and pigment derivatives [55].
- Iron. Iron acquisition is essential for biological processes such as photosynthesis, respiration and nitrogen fixation. Bacteria produce and excrete siderophores, which scavenge iron. Diatoms are not known to produce siderophores, but genome sequence analyses identified the presence of a gene orthologue of a bacterial ferrichrome binding protein that suggests the possibility of iron (III)-siderophore utilization by P. tricornutum [56,57]. Furthermore, it was shown that P. tricornutum was able to uptake siderophores ferrioxamines B and E [58].
- Vitamins. Prokaryotes are thought to be the main producers of B vitamins [59,60]. Although P. tricornutum does not require cobalamin, thiamine and biotin [61], production of organic compounds such as EPA can be considerably enhanced by the bioavailability of co-factors such as cobalamin [62]. This provides the basis for potential mutualistic interactions. For example, Alteromonadales, dominant in our cultures, are thought to be capable of producing B vitamins [63].
- Dissolved Organic Carbon (DOC). It is estimated that up to 50% of carbon fixed via phytoplankton-mediated photosynthesis is utilized by marine bacteria [64], mainly as DOC compounds, defined as the organic material <m in size [65]. DOC from diatoms originates either from live cells or recently lysed or grazed cells, which determines the type of DOCs available, and therefore are likely to influence the bacterial consortia associated with the diatom [30].
- Dissolved Organic Phosphate (DOP). Both diatoms and bacteria primarily utilize orthophosphate as a source of phosphorus. However, to access phosphate from DOP compounds, both diatoms and bacteria developed mechanisms to release orthophosphate (PO) from DOP. The mechanism is not species-specific, which consequently means the “free” orthophosphates can be acquired by any organism [66].
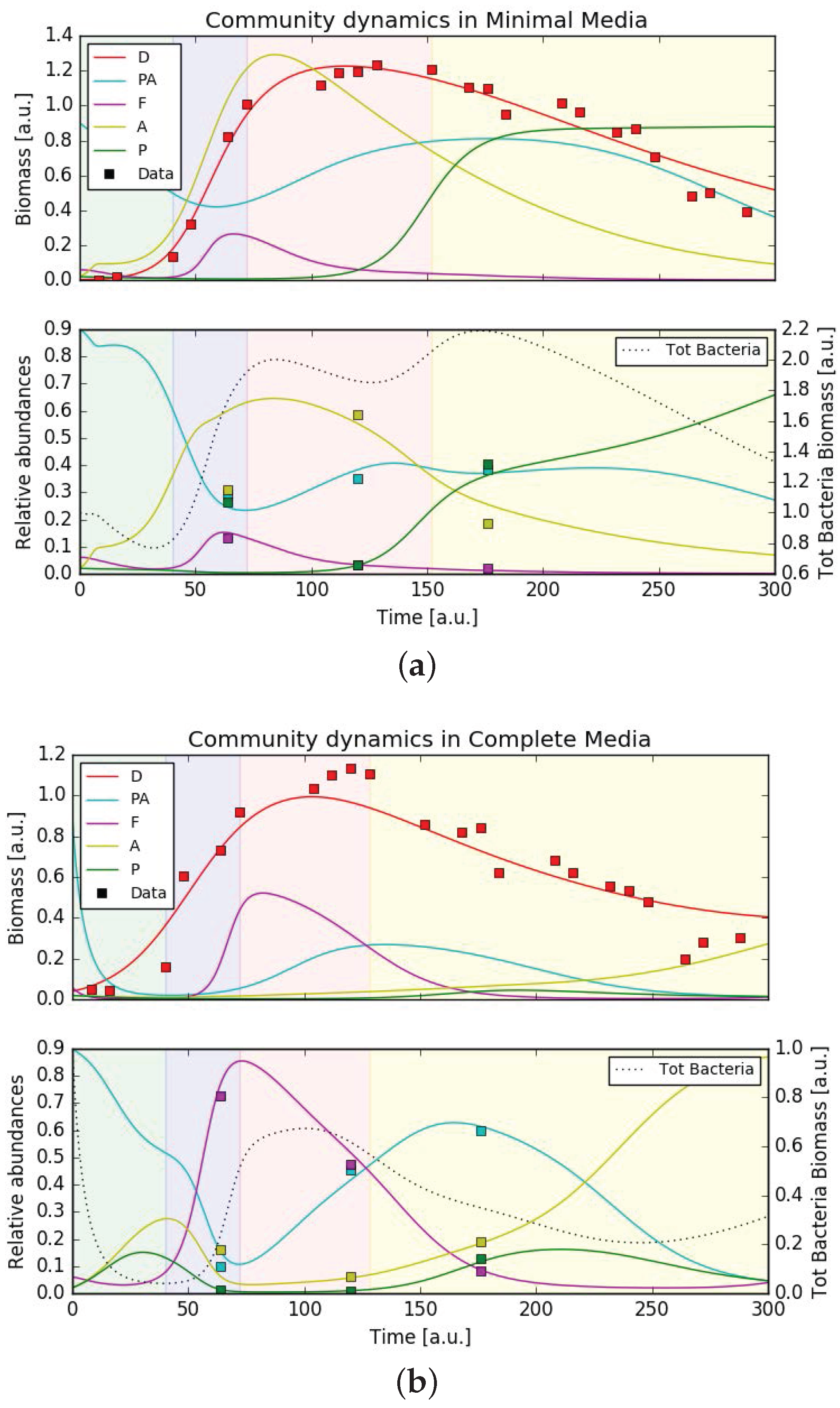
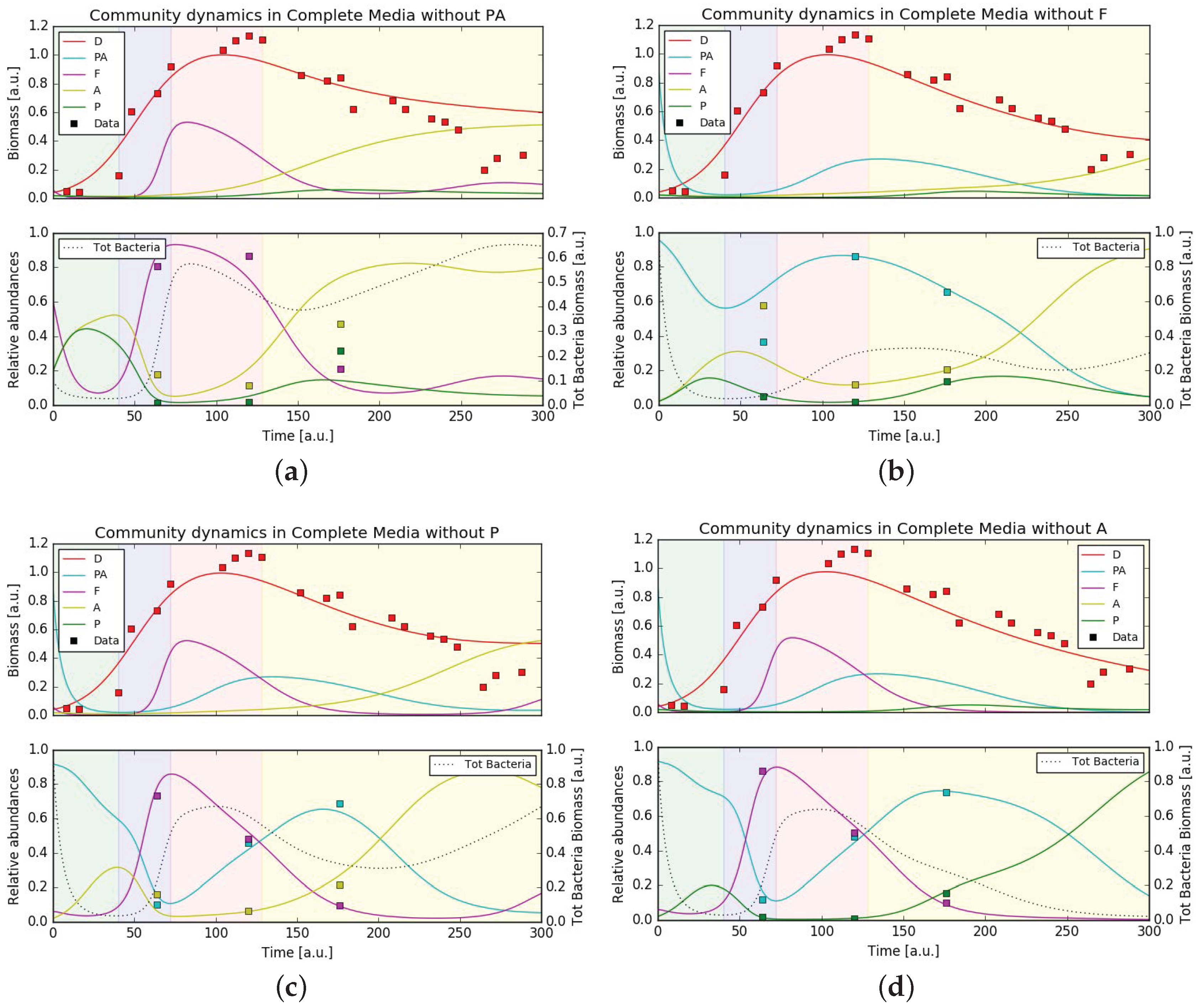
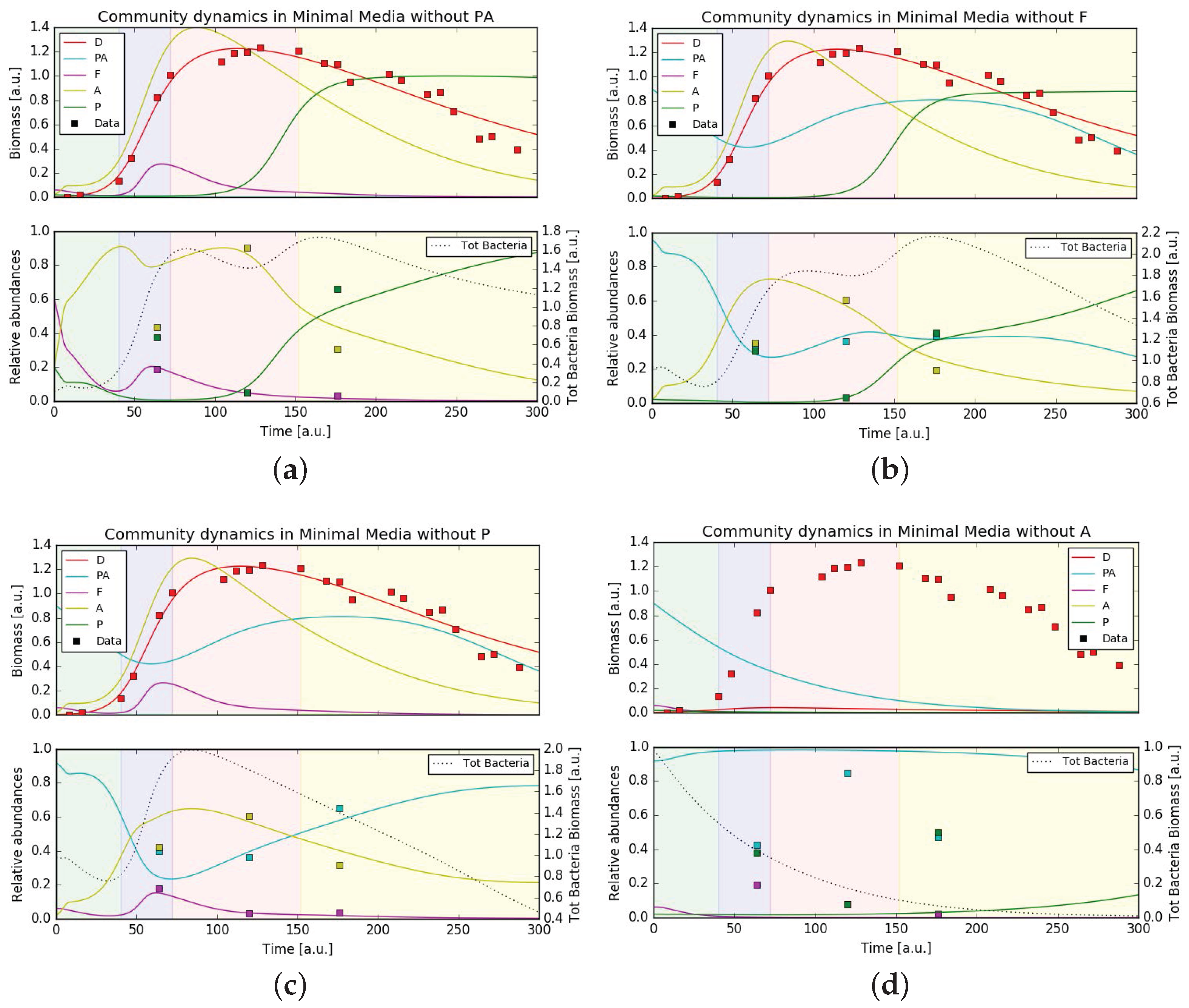
3.5. Mathematical Model Simulations
4. Discussion
4.1. Experimental Observation of the Dynamics of the Bacterial Community Associated to Phaeodactylum tricornutum
4.2. Literature-Based Assessment of the Putative Role of Each Bacterial Family
4.3. Putative Network of Interactions and Validation with a Qualitative Mathematical Model
5. Conclusions and Outlook
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
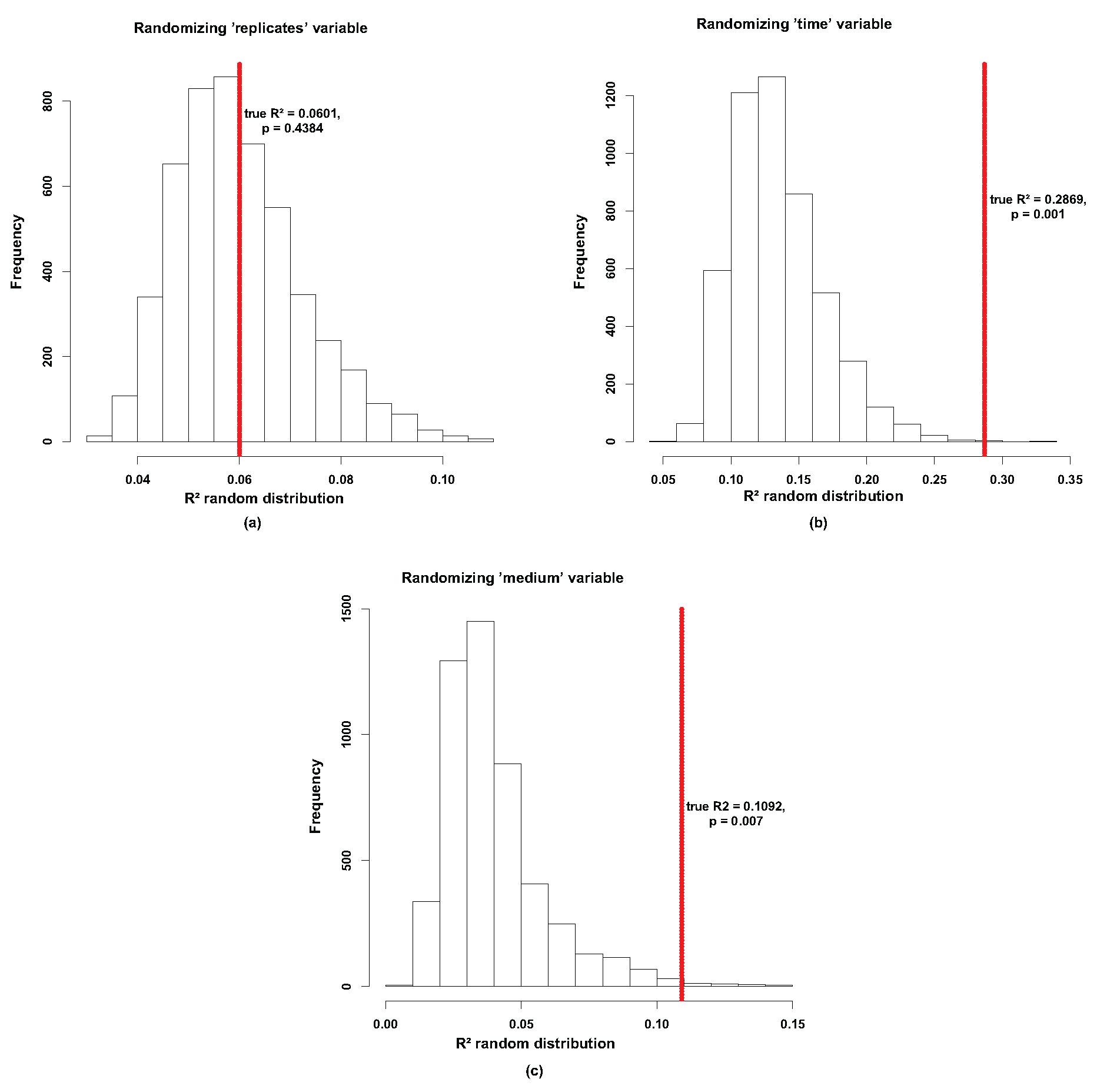
Appendix A. Data Analysis

Appendix B. Mathematical Model Additional Material
- P. tricornutum (D);
- Pseudoalteromonadaceae ();
- Flavobacteriaceae (F);
- Alteromonadaceae (A);
- Pseudomonadaceae (P);
- the dissolved organic carbons of preference for and A ( and , respectively);
- the complex polymers () consumed by F;
- generic vitamins () and iron () needed by D and produced by A;
- bactericidial molecules ( and , produced by D and by respectively);
- the dissolved organic matter ().
- 5 carrying capacities ;
- 34 maximal rates v;
- 15 Monod-type coefficients K;
- the fraction of -dependent growth of A, .
Appendix B.1. ODEs System
Appendix B.1.1. and COP Production

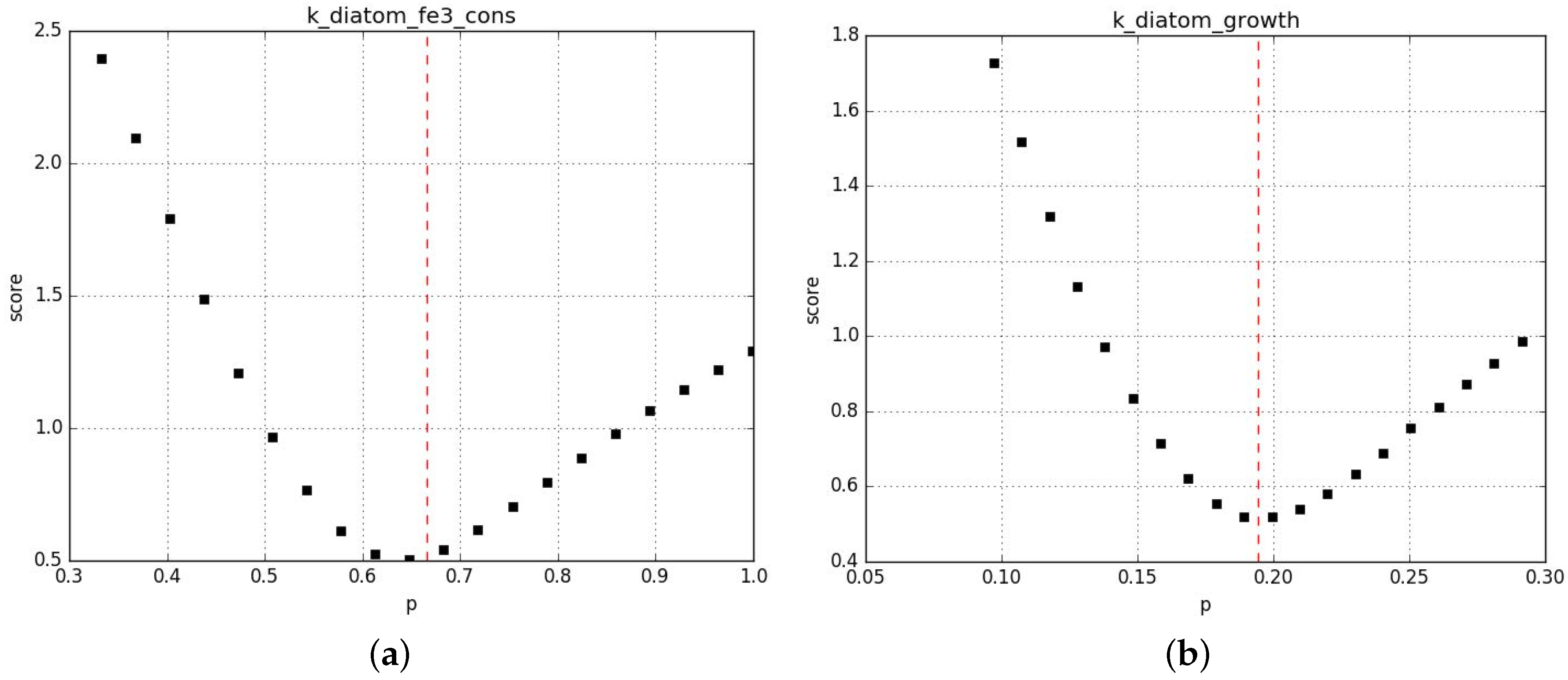
Appendix B.2. Parameter Fitting

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter Sub-Set | Degradation | |||||
|---|---|---|---|---|---|---|
| Sub-set size | 15 | 9 | 14 | 8 | 8 | 2 |
- (1)
- the first generation is populated by extracting the parameters from random uniform distributions within user-chosen ranges;
- (2)
- for each the ODE system is solved and a fitness score (see Appendix B.2.1) is computed;
- (3)
- the most fit 10% individuals are retained as parents for the next generation;
- (4)
- the remaining individuals have a probability to be also selected as parents;
- (5)
- parents are crossed to obtain enough children to reach the original population size;
- (6)
- crossing means randomly pick a parameter sub-set from one parent or the other;
- (7)
- each children has a probability to randomly mutate one parameter;
- (8)
- the process is repeated from step 2. until generation .
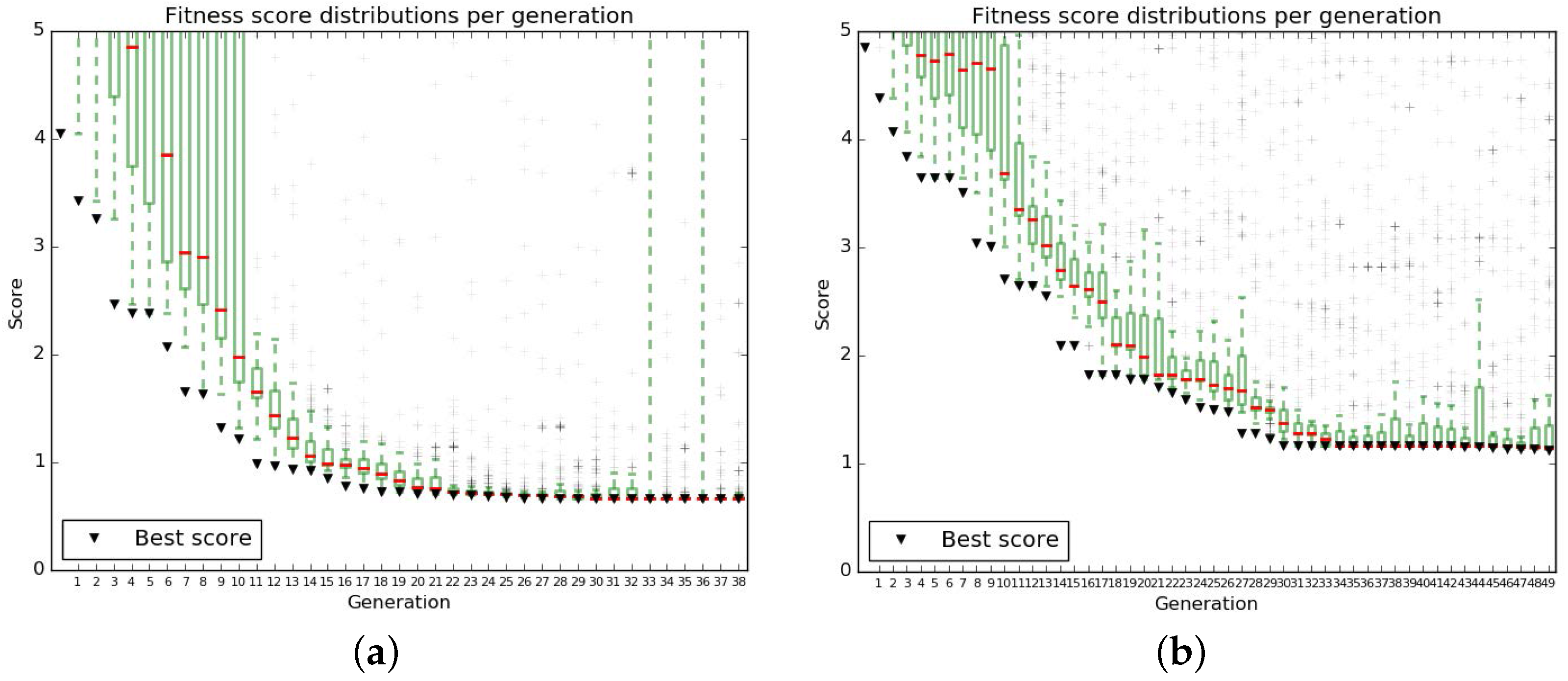
Appendix B.2.1. Fitness Score
| T | 8 | 16 | 40 | 48 | 64 | 72 | 104 | 112 | 120 | 128 | 152 |
| MM | 0.004 | 0.021 | 0.133 | 0.325 | 0.820 | 1.012 | 1.121 | 1.187 | 1.192 | 1.233 | 1.209 |
| CM | 0.050 | 0.044 | 0.162 | 0.605 | 0.733 | 0.919 | 1.037 | 1.099 | 1.134 | 1.108 | 0.859 |
| T | 168 | 176 | 184 | 208 | 216 | 232 | 240 | 248 | 264 | 272 | 288 |
| MM | 1.104 | 1.096 | 0.951 | 1.015 | 0.965 | 0.851 | 0.869 | 0.704 | 0.481 | 0.504 | 0.394 |
| CM | 0.821 | 0.844 | 0.624 | 0.682 | 0.624 | 0.556 | 0.535 | 0.478 | 0.199 | 0.282 | 0.303 |
| Complete Media | Minimal Media | |||||||
|---|---|---|---|---|---|---|---|---|
| t | PA | F | A | P | PA | F | A | P |
| 64 | 0.101 | 0.724 | 0.159 | 0.014 | 0.294 | 0.132 | 0.308 | 0.264 |
| 120 | 0.453 | 0.474 | 0.061 | 0.010 | 0.351 | 0.031 | 0.585 | 0.031 |
| 176 | 0.600 | 0.084 | 0.189 | 0.126 | 0.385 | 0.020 | 0.187 | 0.406 |
Appendix B.2.2. Results of the Genetic Algorithm
- D-MM: D Biomass in Minimal Media;
- D-CM: D Biomass in Complete Media;
- B-MM: Bacteria relative abundances in Minimal Media;
- B-CM: Bacteria relative abundances in Complete Media;
- D*B-MM: D Biomass and Bacteria relative abundances in Minimal Media;
- D*B-CM: D Biomass and Bacteria relative abundances in Complete Media;
- B-fit is run 20 times varying all 55 parameters in ranges
- The parameters from the best B-fits are kept ( and )
- After checking the effect of varying the different parameters sets, different variation ranges are chosen to perform refits
- D*B-CM is run 5 times varying %, %, %
- D*B-MM is run 5 times varying %, and the best parameters are kept ()
- D*B-MM is run again 5 times varying %, %
| 0.562780 | 0.476829 | 0.235751 | 0.23821 | 0.281329 | 0.257813 | |
| 0.463690 | 0.249183 | 0.152283 | 0.02253 | 0.332969 | 0.365116 | |
| 1.043490 | 0.526842 | 0.339699 | 0.82552 | 0.671145 | 0.475618 | |
| 1.884690 | 1.433304 | 0.495578 | 0.94702 | 1.085873 | 0.889537 | |
| 1.230920 | 1.112280 | 0.351905 | 2.74047 | 1.564099 | 0.668637 | |
| 0.036310 | 0.067159 | 0.085177 | 0.01697 | 0.058289 | 0.113161 | |
| 0.504220 | 0.504324 | 0.149340 | 1.30073 | 0.806015 | 0.315439 | |
| 0.204470 | 0.464587 | 0.226651 | 0.99257 | 0.670187 | 0.250496 | |
| 0.186340 | 0.493623 | 0.298676 | 0.74598 | 0.349684 | 0.246408 | |
| 0.795530 | 0.508826 | 0.229097 | 0.00743 | 0.323377 | 0.248408 | |
| 0.030470 | 0.052871 | 0.094576 | 0.06702 | 0.130514 | 0.176652 | |
| 0.134290 | 0.118939 | 0.028298 | 0.19948 | 0.206115 | 0.091565 | |
| 0.329520 | 0.434683 | 0.340005 | 0.34841 | 1.037434 | 0.510106 | |
| 0.954240 | 0.644251 | 0.317607 | 1.09226 | 1.241136 | 0.618984 | |
| 0.108110 | 0.409152 | 0.213390 | 0.07769 | 0.263422 | 0.330743 | |
| 0.350050 | 0.373353 | 0.117858 | 0.57995 | 0.784058 | 0.412260 | |
| 0.488680 | 0.583597 | 0.358354 | 0.02979 | 0.124157 | 0.134217 | |
| 1.199730 | 1.048777 | 0.343732 | 0.33321 | 0.486298 | 0.256807 | |
| 0.844900 | 0.645544 | 0.226955 | 0.46274 | 0.346839 | 0.088571 | |
| 0.469600 | 0.903463 | 0.501364 | 0.09782 | 0.339058 | 0.356208 | |
| 0.314330 | 0.853140 | 0.557108 | 0.36480 | 0.546552 | 0.330353 | |
| 1.875200 | 1.584920 | 0.515701 | 1.57897 | 1.427444 | 0.420945 | |
| 1.005110 | 1.268507 | 0.463020 | 0.70666 | 0.736754 | 0.370734 | |
| 0.007180 | 0.016960 | 0.051891 | 0.00681 | 0.013765 | 0.049375 | |
| 1.770740 | 0.987437 | 0.490546 | 1.65657 | 1.350378 | 0.421454 | |
| 1.055270 | 0.990547 | 0.415673 | 0.83897 | 1.081959 | 0.591094 | |
| 0.135980 | 0.653181 | 0.351953 | 0.54133 | 0.565364 | 0.231813 | |
| 1.214350 | 0.899207 | 0.360058 | 1.28659 | 1.070999 | 0.478554 | |
| 0.665740 | 0.755699 | 0.241111 | 0.31684 | 0.363974 | 0.099207 | |
| 0.194310 | 0.200395 | 0.069030 | 0.52737 | 0.562459 | 0.149546 | |
| 0.367880 | 0.566566 | 0.416514 | 1.78450 | 0.909564 | 0.404000 | |
| 0.936420 | 0.583317 | 0.263447 | 0.16731 | 0.299761 | 0.198921 | |
| 0.477700 | 0.588674 | 0.234155 | 0.74525 | 0.451922 | 0.315203 | |
| 0.184360 | 0.311845 | 0.105780 | 0.23234 | 1.237169 | 1.012458 | |
| 1.351050 | 1.206888 | 0.384417 | 0.54187 | 1.074951 | 0.758107 | |
| 0.139320 | 0.175972 | 0.045416 | 0.57005 | 0.330531 | 0.127086 | |
| 0.382820 | 0.318181 | 0.144775 | 0.18005 | 0.200895 | 0.150824 | |
| 0.092860 | 0.080066 | 0.074871 | 0.00984 | 0.135875 | 0.283818 | |
| 0.765450 | 0.726578 | 0.223690 | 1.50156 | 0.888556 | 0.545041 | |
| 0.020100 | 0.148399 | 0.132143 | 0.16823 | 0.326145 | 0.294570 | |
| 0.609800 | 0.560853 | 0.171693 | 1.12080 | 0.688621 | 0.413129 | |
| 1.009740 | 1.238831 | 0.430709 | 2.11081 | 1.419892 | 0.958683 | |
| 1.301320 | 1.277678 | 0.407513 | 1.17750 | 2.585117 | 0.869802 | |
| 0.020440 | 0.069033 | 0.167148 | 0.01591 | 0.036821 | 0.150349 | |
| 0.698330 | 0.523151 | 0.203572 | 0.17625 | 0.124345 | 0.136063 | |
| 0.195450 | 0.189091 | 0.103414 | 0.03107 | 0.116528 | 0.203261 | |
| 0.820720 | 0.440066 | 0.249502 | 0.57938 | 0.527859 | 0.284980 | |
| 0.245720 | 0.351941 | 0.221873 | 0.42128 | 0.564689 | 0.489764 | |
| 0.755570 | 0.541606 | 0.297743 | 0.05329 | 0.404319 | 0.414359 | |
| 1.577050 | 1.484135 | 0.474372 | 2.65508 | 1.368551 | 0.892632 | |
| 0.819580 | 0.848959 | 0.264185 | 0.28618 | 0.568550 | 0.438164 | |
| 0.995130 | 1.029216 | 0.323852 | 1.28138 | 1.477045 | 0.533872 | |
| 0.221040 | 0.284309 | 0.181709 | 0.01861 | 0.052638 | 0.102986 | |
| 0.236820 | 0.254966 | 0.144860 | 0.41130 | 0.249994 | 0.182493 | |
| 0.130620 | 0.110548 | 0.125387 | 0.01816 | 0.108930 | 0.154334 | |
| 0.327430 | 0.468045 | 0.287832 | 0.12769 | 0.350329 | 0.210662 |
Appendix B.2.3. Sanity Checks of the Parameter Fits





References
- Chisti, Y. Biodiesel from microalgae. Biotechnol. Adv. 2007, 25, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Moejes, F.W.; Moejes, K.B. Algae for Africa: Microalgae as a source of food, feed and fuel in Kenya. Afr. J.Biotechnol. 2017, 16, 288–301. [Google Scholar] [CrossRef]
- Ryther, J.; Goldman, J. Microbes as food in mariculture. Annu. Rev. Microbiol. 1975, 29, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Tredici, M.R.; Biondi, N.; Ponis, E.; Rodolfi, L.; Zittelli, G.C.; Burnell, G.; Allan, G. Advances in microalgal culture for aquaculture feed and other uses. In New Technologies in Aquaculture: Improving Production Efficiency, Quality and Environmental Management; Burnell, G., Allan, G., Eds.; Woodhead Publishing Ltd.: Cambridge, UK, 2009; pp. 610–676. [Google Scholar]
- Rebolloso-Fuentes, M.; Navarro-Pérez, A.; Ramos-Miras, J.; Guil-Guerrero, J. Biomass nutrient profiles of the microalga Phaeodactylum tricornutum. J. Food Biochem. 2001, 25, 57–76. [Google Scholar] [CrossRef]
- Kates, M.; Volcani, B.E. Lipid components of diatoms. Biochim. Biophys. Acta 1966, 116, 264–278. [Google Scholar] [CrossRef]
- Fajardo, A.R.; Cerdán, L.E.; Medina, A.R.; Fernández, F.G.A.; Moreno, P.A.G.; Grima, E.M. Lipid extraction from the microalga Phaeodactylum tricornutum. Eur. J. Lipid Sci. Technol. 2007, 109, 120–126. [Google Scholar] [CrossRef]
- Siron, R.; Giusti, G.; Berland, B. Changes in the fatty acid composition of Phaeodactylum tricornutum and Dunaliella tertiolecta during growth and under phosphorus deficiency. Mar. Ecol. Prog. Ser. 1989, 55, 95–100. [Google Scholar] [CrossRef]
- Yashodhara, B.M.; Umakanth, S.; Pappachan, J.M.; Bhat, S.K.; Kamath, R.; Choo, B.H. Omega-3 fatty acids: A comprehensive review of their role in health and disease. Postgrad. Med. J. 2009, 85, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Francius, G.; Tesson, B.; Dague, E.; Martin-jézéquel, V.; Dufrêne, Y.F. Nanostructure and nanomechanics of live Phaeodactylum tricornutum morphotypes. Environ. Microbiol. 2008, 10, 1344–1356. [Google Scholar] [CrossRef] [PubMed]
- Santaeufemia, S.; Torres, E.; Mera, R.; Abalde, J. Bioremediation of oxytetracycline in seawater by living and dead biomass of the microalga Phaeodactylum tricornutum. J. Hazard. Mater. 2016, 320, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Mata, T.; Martins, A.; Caetano, N. Microalgae for biodiesel production and other applications: A review. Renew. Sustain. Energy Rev. 2010, 14, 217–232. [Google Scholar] [CrossRef]
- Grima, E.; Fernández, F. Photobioreactors: Light regime, mass transfer, and scaleup. J. Biotechnol. 1999, 70, 231–247. [Google Scholar] [CrossRef]
- Parmar, A.; Singh, N.; Pandey, A. Cyanobacteria and microalgae: A positive prospect for biofuels. Bioresour. Technol. 2011, 102, 10163–10172. [Google Scholar] [CrossRef] [PubMed]
- Day, J.G.; Thomas, N.J.; Achilles-Day, U.E.M.; Leakey, R.J.G. Early detection of protozoan grazers in algal biofuel cultures. Bioresour. Technol. 2012, 114, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, W.; Chen, L.; Wang, J.; Liu, T. The contamination and control of biological pollutants in mass cultivation of microalgae. Bioresour. Technol. 2013, 128, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Kazamia, E.; Aldridge, D.C.; Smith, A.G. Synthetic ecology—A way forward for sustainable algal biofuel production? J. Biotechnol. 2012, 162, 163–169. [Google Scholar] [CrossRef]
- Gause, G. The Struggle for Existence; The Williams & Wilkins company: Baltimore, MD, USA, 1934; p. 163. [Google Scholar]
- Hardin, G. The competitive exclusion principle. Science 1960, 131, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Dykhuizen, D. Santa Rosalia revisited: Why are there so many species of bacteria? Antonie Van Leeuwenhoek 1998, 73, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, G.I.; Levin, S.A. Marine Ecosystems as Complex Adaptive Systems: Emergent Patterns, Critical Transitions, and Public Goods. Ecosystems 2017, 20, 458–476. [Google Scholar] [CrossRef]
- Widder, S.; Allen, R.J.; Pfeiffer, T.; Curtis, T.P.; Wiuf, C.; Sloan, W.T.; Cordero, O.X.; Brown, S.P.; Momeni, B.; Shou, W.; et al. Challenges in microbial ecology: Building predictive understanding of community function and dynamics. ISME J. 2016, 10, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Song, H.S.; Cannon, W.; Beliaev, A.; Konopka, A. Mathematical Modeling of Microbial Community Dynamics: A Methodological Review. Processes 2014, 2, 711–752. [Google Scholar] [CrossRef]
- Zomorrodi, A.R.; Segrè, D. Synthetic Ecology of Microbes: Mathematical Models and Applications. J. Mol. Biol. 2015, 428, 837–861. [Google Scholar] [CrossRef] [PubMed]
- Succurro, A.; Moejes, F.W.; Ebenhöh, O. A Diverse Community To Study Communities: Integration of Experiments and Mathematical Models To Study Microbial Consortia. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef] [PubMed]
- Lotka, A. Elements of Physical Biology; Williams & Wilkins Company: Philadelphia, PA, USA, 1925. [Google Scholar]
- Volterra, V. Fluctuations in the abundance of a species considered mathematically. Nature 1926, 118, 558–560. [Google Scholar] [CrossRef]
- Hofbauer, J.; Hutson, V.; Jansen, W. Coexistence for systems governed by difference equations of Lotka-Volterra type. J. Math. Biol. 1987, 25, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Parker, M.S.; Armbrust, E.V. Interactions between diatoms and bacteria. Microbiol. Mol. Biol. Rev. 2012, 76, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Provasoli, L. Nutrition and ecology of Protozoa and Algae. Annu. Rev. Microbiol. 1958, 12, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Delucca, R.; McCracken, M.D. Observations on interactions between naturally-collected bacteria and several species of algae. Hydrobiologia 1977, 55, 71–75. [Google Scholar] [CrossRef]
- Suminto; Hirayama, K. Application of a growth-promoting bacteria for stable mass culture of three marine microalgae. Hydrobiologia 1997, 358, 223–230. [Google Scholar]
- Bruckner, C.G.; Rehm, C.; Grossart, H.P.; Kroth, P.G. Growth and release of extracellular organic compounds by benthic diatoms depend on interactions with bacteria. Environ. Microbiol. 2011, 13, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Hmelo, L.R.; van Tol, H.M.; Durham, B.P.; Carlson, L.T.; Heal, K.R.; Morales, R.L.; Berthiaume, C.T.; Parker, M.S.; Djunaedi, B.; et al. Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 2015, 522, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Verhulst, P. Notice sur la loi que la population suit dans son accroissement. Corresp. Math. Phys. l’Obs. Brux. 1838, 10, 113–121. [Google Scholar]
- Gachon, C.M.; Day, J.G.; Campbell, C.N.; Pröschold, T.; Saxon, R.J.; Küpper, F.C. The Culture Collection of Algae and Protozoa (CCAP): A biological resource for protistan genomics. Gene 2007, 406, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Moejes, F.W. Dynamics of the bacterial community associated with Phaeodactylum tricornutum cultures: A novel approach to scaling up microalgal cultures. Ph.D. Thesis, Heinrich Heine University Dusseldorf, Düsseldorf, Germany, 2016. [Google Scholar]
- Guillard, R.; Ryther, J. Studies of marine planktonic diatoms: I. Cyclotella nana Hutedt, and Detonula confervacea (Cleve) Gran. Can. J. Microbiol. 1962, 8, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Guillard, R. Culture of phytoplankton for feeding marine invertebrates. In Culture of Marine Invertebrate Animals; Smith, W.L., Chanley, M.H., Eds.; Plenum Press: New York, NY, USA, 1975; pp. 29–60. [Google Scholar]
- De Gouvion Saint Cyr, D.; Wisniewski, C.; Schrive, L.; Farhi, E.; Rivasseau, C. Feasibility study of microfiltration for algae separation in an innovative nuclear effluents decontamination process. Sep. Purif. Technol. 2014, 125, 126–135. [Google Scholar] [CrossRef]
- Quail, M.; Smith, M.E.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion torrent, pacific biosciences and illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- Grada, A.; Weinbrecht, K. Next-Generation Sequencing: Methodology and Application. J. Investig. Dermatol. 2013, 133, e11. [Google Scholar] [CrossRef] [PubMed]
- Pylro, V.S.; Roesch, L.F.W.; Morais, D.K.; Clark, I.M.; Hirsch, P.R.; Tótola, M.R. Data analysis for 16S microbial profiling from different benchtop sequencing platforms. J. Microbiol. Methods 2014, 107, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Aronesty, E. ea-utils: Command-Line Tools for Processing Biological Sequencing Data. Durham, NC, USA, 2011. Available online: https://github.com/ExpressionAnalysis/ea-utils (accessed on 30 November 2017).
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2015. Available online: http://www.R-project.org/ (accessed on 30 November 2017).
- Monod, J. The Growth of Bacterial Cultures. Annu. Rev. Microbiol. 1949, 3, 371–394. [Google Scholar] [CrossRef]
- Mitchell, M. An Introduction to Genetic Algorithms; MIT Press: Cambridge, MA, USA, 1996. [Google Scholar]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J. Bioactive compound synthetic capacity and ecological significance of marine bacterial genus Pseudoalteromonas. Mar. Drugs 2007, 5, 220–241. [Google Scholar] [CrossRef] [PubMed]
- Desbois, A.P.; Mearns-Spragg, A.; Smith, V.J. A Fatty Acid from the Diatom Phaeodactylum tricornutum is Antibacterial Against Diverse Bacteria Including Multi-resistant Staphylococcus aureus (MRSA). Mar. Biotechnol. 2009, 11, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Soria-Dengg, S.; Reissbrodt, R.; Horstmann, U. Siderophores in marine coastal waters and their relevance for iron uptake by phytoplankton: Experiments with the diatom Phaeodactylum tricornutum. Mar. Ecol. Prog. Ser. 2001, 220, 73–82. [Google Scholar] [CrossRef]
- Amin, S.A.; Green, D.H.; Hart, M.C.; Küpper, F.C.; Sunda, W.G.; Carrano, C.J. Photolysis of iron‚ siderophore chelates promotes bacterial‚ algal mutualism. Proc. Natl. Acad. Sci. USA 2009, 106, 17071–17076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria-Dengg, S.; Horstmann, U. Ferrioxamines B and E as iron sources for the marine diatom Phaeodactylum tricornutum. Mar. Ecol. Prog. Ser. 1995, 127, 269–277. [Google Scholar] [CrossRef]
- Provasoli, L. Organic regulation of phytoplankton fertility. In The Sea: Ideas and Observations on Progress in the Study of the Seas; Hill, M., Ed.; Wiley-Interscience: New York, NY, USA, 1963; pp. 165–219. [Google Scholar]
- Provasoli, L.; Carlucci, A. Vitamins and growth regulators. In Algal Physiology and Biochemistry, Botanical Monographs, 10; Stewart, W., Ed.; Blackwell Scientific Publications: San Diego, CA, USA, 1974; pp. 741–787. [Google Scholar]
- Croft, M.T.; Warren, M.J.; Smith, A.G. Algae need their vitamins. Eukaryot. Cell 2006, 5, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Yongmanitchai, W.; Ward, O.P. Growth of and omega-3 fatty acid production by Phaeodactylum tricornutum under different culture conditions. Appl. Environ. Microbiol. 1991, 57, 419–425. [Google Scholar] [PubMed]
- Sañudo-Wilhelmy, S.A.; Gómez-Consarnau, L.; Suffridge, C.; Webb, E.A. The Role of B Vitamins in Marine Biogeochemistry. Annu. Rev. Mar. Sci. 2014, 6, 339–367. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Fenchel, T.; Field, J.; Gray, J.; Meyer-Reil, L.; Thingstad, F. The Ecological Role of Water-Column Microbes in the Sea. Mar. Ecol. Prog. Ser. 1983, 10, 257–263. [Google Scholar] [CrossRef]
- Stocker, R. Marine Microbes See a Sea of Gradients. Science 2012, 338, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Persson, G.; Jansson, M.; Kluwer, C. Phosphate uptake and utilizaton by bacteria and algae. Hydrobiologia 1988, 170, 177–189. [Google Scholar]
- Bell, W.; Mitchell, R. Chemotactic and growth responses of marine bacteria to algal extracellular products. Biol. Bull. 1972, 143, 265–277. [Google Scholar] [CrossRef]
- MacArthur, R. Fluctuations of animal populations and a measure of community stability. Ecology 1955, 36, 533–536. [Google Scholar] [CrossRef]
- Gardner, M.; Ashby, W. Connectance of large dynamic (cybernetic) systems: Critical values for stability. Nature 1970, 228, 784. [Google Scholar] [CrossRef] [PubMed]
- Pimm, S. The complexity and stability of ecosystems. Nature 1984, 307, 321–326. [Google Scholar] [CrossRef]
- Elton, C. The Ecology of Invasions by Animals and Plants; Springer: Dordrecht, The Netherlands, 1958. [Google Scholar]
- McCann, K.S. The diversity-stability debate. Nature 2000, 405, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, E.P.; Flavier, S.; Christen, R. Phylogenetic relationships among marine Alteromonas-like proteobacteria: Emended description of the family Alteromonadaceae and proposal of Pseudoalteromonadaceae fam. nov., Colwelliaceae fam. nov., Shewanellaceae fam. nov., Moritellaceae fam. nov., Ferri. Int. J. Syst. Evolut. Microbiol. 2004, 54, 1773–1788. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.; DeLong, E.F.; Lory, S.; Stackebrandt, E.; Thompson, F. The Prokaryotes; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Lee, S.O.; Kato, J.; Takiguchi, N.; Kuroda, A.; Ikeda, T. Involvement of an Extracellular Protease in Algicidal Activity of the Marine Bacterium Pseudoalteromonas sp. Strain A28. Appl. Environ. Microbiol. 2000, 66, 4334–4339. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, K.; Dohmoto, N. Pseudoalteromonas peptidolytica sp. nov., a novel marine mussel-thread-degrading bacterium isolated from the Sea of Japan. Int. J. Syst. Evolut. Microbiol. 2000, 50, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xie, B.; Lu, J.; He, H.; Zhang, Y. A novel type of subtilase from the psychrotolerant bacterium Pseudoalteromonas sp. SM9913: Catalytic and structural properties of deseasin MCP-01. Microbiology 2007, 153, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Khudary, R.A.; Venkatachalam, R.; Katzer, M. A cold-adapted esterase of a novel marine isolate, Pseudoalteromonas arctica: Gene cloning, enzyme purification and characterization. Extremophiles 2010, 14, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Wang, S.; Fang, Y.; Li, H.; Liu, S.; Liu, H. Cloning, expression, purification, and characterization of cold-adapted α-amylase from Pseudoalteromonas arctica GS230. Protein J. 2010, 29, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Albino, A.; Marco, S.; Maro, A.D. Characterization of a cold-adapted glutathione synthetase from the psychrophile Pseudoalteromonas haloplanktis. Mol. Biosyst. 2012, 8, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Guo, J.; Chen, X.; Xie, B.; Zhang, X. Structural and functional characterization of mature forms of metalloprotease E495 from Arctic sea-ice bacterium Pseudoalteromonas sp. SM495. PLoS ONE 2012, 7, e35442. [Google Scholar] [CrossRef] [PubMed]
- Holmström, C.; Kjelleberg, S. Marine Pseudoalteromonas species are associated with higher organisms and produce biologically active extracellular agents. FEMS Microbiol. Ecol. 1999, 30, 285–293. [Google Scholar] [CrossRef]
- Ivanova, E.P.; Mikhaĭlov, V.V. A new family of Alteromonadaceae fam. nov., including the marine proteobacteria species Alteromonas, Pseudoalteromonas, Idiomarina i Colwellia. Mikrobiologiia 2001, 70, 15–23. [Google Scholar] [PubMed]
- Reid, R.; Butler, A. Investigation of the mechanism of iron acquisition by the marine bacterium Alteromonas luteoviolaceus: Characterization of siderophore production. Limnol. Oceanogr. 1991, 36, 1783–1792. [Google Scholar] [CrossRef]
- Holt, P.D.; Reid, R.R.; Lewis, B.L.; Luther, G.W.; Butler, A. Iron(III) coordination chemistry of alterobactin A: A siderophore from the marine bacterium Alteromonas luteoviolacea. Inorg. Chem. 2005, 44, 7671–7677. [Google Scholar] [CrossRef] [PubMed]
- Vraspir, J.M.; Butler, A. Chemistry of marine ligands and siderophores. Annu. Rev. Mar. Sci. 2009, 1, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Bruland, K.W.; Donat, J.R.; Hutchins, D.a. Interactive influences of bioactive trace metals on biological production in oceanic waters. Limnol. Oceanogr. 1991, 36, 1555–1577. [Google Scholar] [CrossRef]
- Morel, F.M.M.; Price, N.M. The biogeochemical cycles of trace metals in the oceans. Science 2003, 300, 944–947. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Jo, Y.; Kim, G.J.; Choi, H. Gramella lutea sp. nov., a Novel Species of the Family Flavobacteriaceae Isolated from Marine Sediment. Curr. Microbiol. 2015, 71, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Glöckner, F.O.; Fuchs, B.M.; Amann, R. Bacterioplankton compositions of lakes and oceans: A first comparison based on fluorescence in situ hybridization. Appl. Environ. Microbiol. 1999, 65, 3721–3726. [Google Scholar] [PubMed]
- Abell, G.; Bowman, J. Ecological and biogeographic relationships of class Flavobacteria in the Southern Ocean. FEMS Microbiol. Ecol. 2005, 51, 265–277. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.F.; Preston, C.M.; Mincer, T.; Rich, V.; Hallam, S.J.; Frigaard, N.U.; Martinez, A.; Sullivan, M.B.; Edwards, R.; Brito, B.R.; et al. Community genomics among stratified microbial assemblages in the ocean’s interior. Science 2006, 311, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Manz, W.; Amann, R.; Ludwig, W.; Vancanneyt, M.; Schleifer, K.H. Application of a suite of 16S rRNA-specific oligonucleotide probes designed to investigate bacteria of the phylum cytophaga-flavobacter-bacteroides in the natural environment. Microbiology 1996, 142, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Kirchman, D.L. The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 2002, 39, 91–100. [Google Scholar] [CrossRef]
- Cottrell, M.T.; Kirchman, D.L. Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl. Environ. Microbiol. 2000, 66, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Pinhassi, J.; Sala, M.M.; Havskum, H.; Peters, F.; Guadayol, Ò.; Malits, A.; Marrasé, C. Changes in bacterioplankton composition under different phytoplankton regimens. Appl. Environ. Microbiol. 2004, 70, 6753–6766. [Google Scholar] [CrossRef] [PubMed]
- Smayda, T. Harmful algal blooms: Their ecophysiology and general relevance to phytoplankton blooms in the sea. Limnol. Oceanogr. 1997, 42, 1137–1153. [Google Scholar] [CrossRef]
- Starr, M.P.; Stolp, H.; Trüper, H.G.; Balows, A.; Schlegel, H.G. The Prokaryotes; Springer: Berlin/Heidelberg, Germany, 1981. [Google Scholar]
- Anzai, Y.; Kim, H.; Park, J.Y.; Wakabayashi, H.; Oyaizu, H. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int. J. Syst. Evolut. Microbiol. 2000, 50, 1563–1589. [Google Scholar] [CrossRef] [PubMed]
- Isnansetyo, A.; Kamei, Y. Bioactive substances produced by marine isolates of Pseudomonas. J. Ind. Microbiol. Biotechnol. 2009, 36, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Zelezniak, A.; Andrejev, S.; Ponomarova, O.; Mende, D.R.; Bork, P.; Patil, K.R. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc. Natl. Acad. Sci. USA 2015, 112, 6449–6454. [Google Scholar] [CrossRef] [PubMed]
- Ponomarova, O.; Patil, K.R. Metabolic interactions in microbial communities: Untangling the Gordian knot. Curr. Opin. Microbiol. 2015, 27, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Bausch, C.; Richmond, C.; Blattner, F.R.; Conway, T. Functional genomics: Expression analysis of Escherichia coli growing on minimal and rich media. J. Bacteriol. 1999, 181, 6425–6440. [Google Scholar] [PubMed]
- Beg, Q.K.; Zampieri, M.; Klitgord, N.; Collins, S.B.; Altafini, C.; Serres, M.H.; Segrè, D. Detection of transcriptional triggers in the dynamics of microbial growth: Application to the respiratorily versatile bacterium Shewanella oneidensis. Nucleic Acids Res. 2012, 40, 7132–7149. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, E.M.; Allen, A.E.; Dupont, C.L.; Norden-Krichmar, T.M.; Bai, J.; Valas, R.E.; Saito, M.A. Influence of cobalamin scarcity on diatom molecular physiology and identification of a cobalamin acquisition protein. Proc. Natl. Acad. Sci. USA 2012, 109, E1762–E1771. [Google Scholar] [CrossRef] [PubMed]
- Horgan, R.P.; Kenny, L.C. ‘Omic’ technologies: Genomics, transcriptomics, proteomics and metabolomics. Obstet. Gynaecol. 2011, 13, 189–195. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moejes, F.W.; Succurro, A.; Popa, O.; Maguire, J.; Ebenhöh, O. Dynamics of the Bacterial Community Associated with Phaeodactylum tricornutum Cultures. Processes 2017, 5, 77. https://doi.org/10.3390/pr5040077
Moejes FW, Succurro A, Popa O, Maguire J, Ebenhöh O. Dynamics of the Bacterial Community Associated with Phaeodactylum tricornutum Cultures. Processes. 2017; 5(4):77. https://doi.org/10.3390/pr5040077
Chicago/Turabian StyleMoejes, Fiona Wanjiku, Antonella Succurro, Ovidiu Popa, Julie Maguire, and Oliver Ebenhöh. 2017. "Dynamics of the Bacterial Community Associated with Phaeodactylum tricornutum Cultures" Processes 5, no. 4: 77. https://doi.org/10.3390/pr5040077
APA StyleMoejes, F. W., Succurro, A., Popa, O., Maguire, J., & Ebenhöh, O. (2017). Dynamics of the Bacterial Community Associated with Phaeodactylum tricornutum Cultures. Processes, 5(4), 77. https://doi.org/10.3390/pr5040077