A Robust Method for the Estimation of Kinetic Parameters for Systems Including Slow and Rapid Reactions—From Differential-Algebraic Model to Differential Model



Abstract
:1. Introduction
2. Development of a Robust Method
- T1.
- Mass balances of all components in the batch reactor are written down (ODEs)
- T2.
- A quasi-steady-state hypothesis is applied to the intermediates—the ODEs become a system of DAEs is created
- T3.
- The DAE system is converted to a set of implicit ODEs by differentiation
- T4.
- The system of implicit ODEs is converted to a set of explicit ODEs
- T5.
- The system of explicit ODEs is implemented in a combined stiff ODE solver—parameter estimation software Modest.
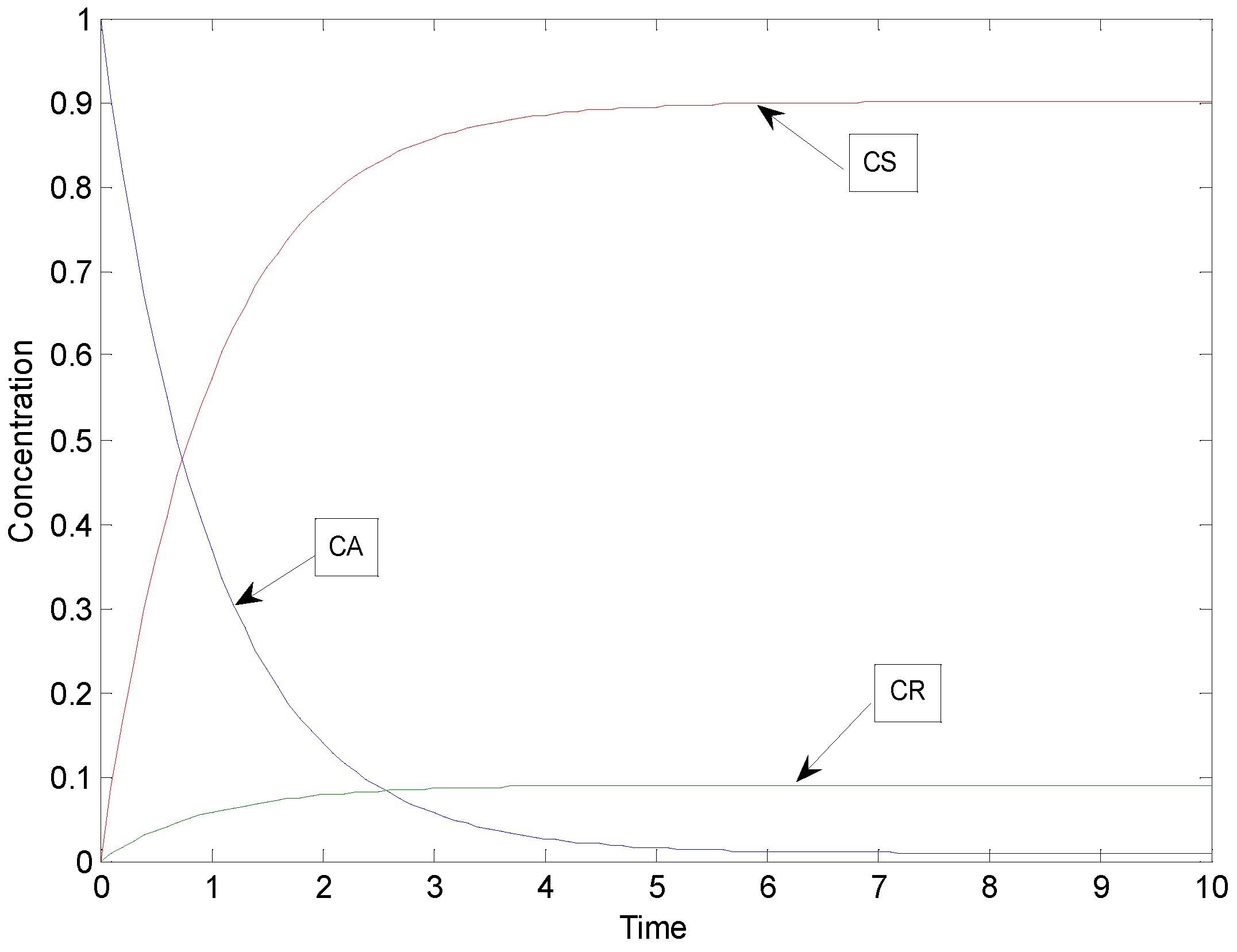
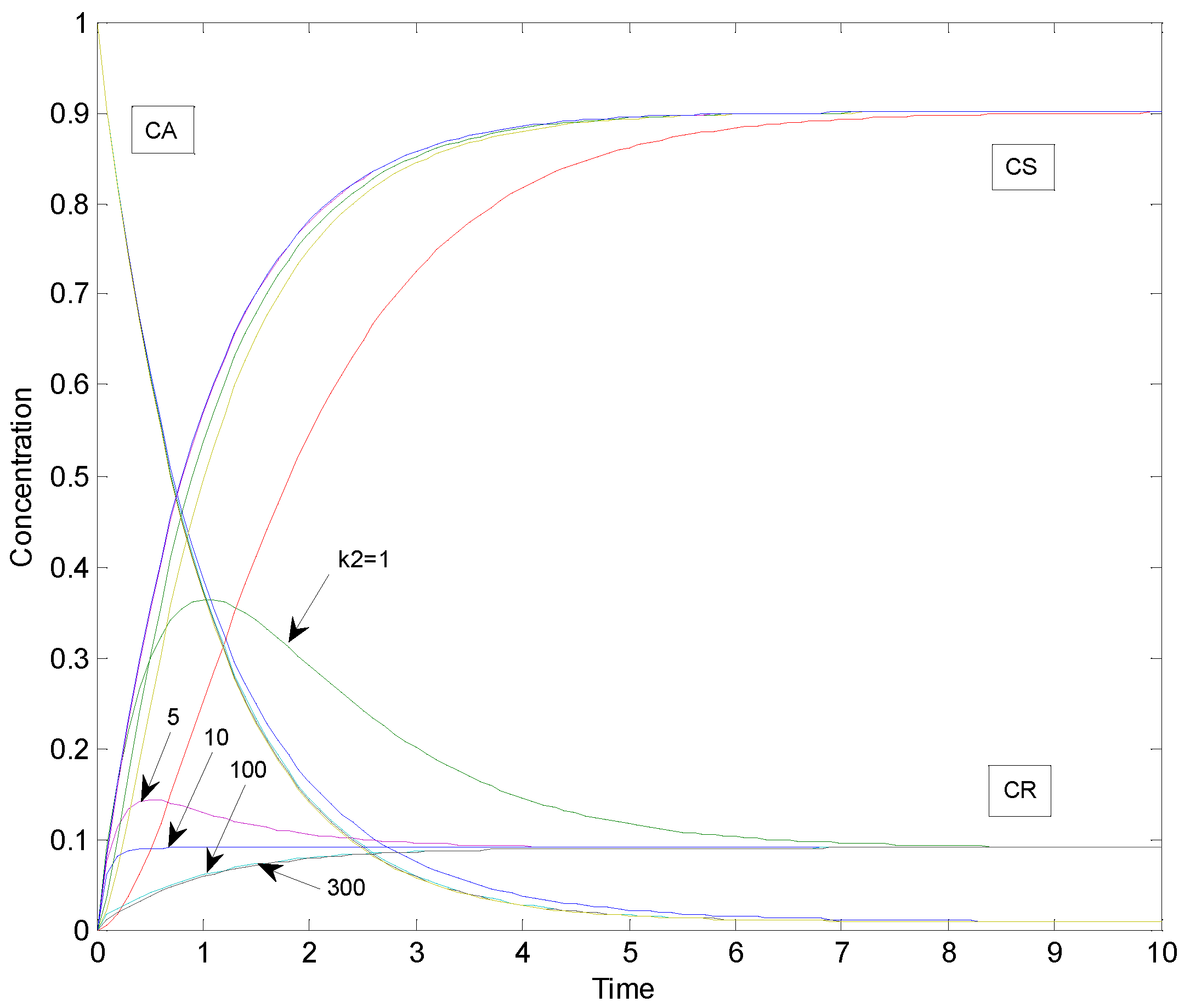
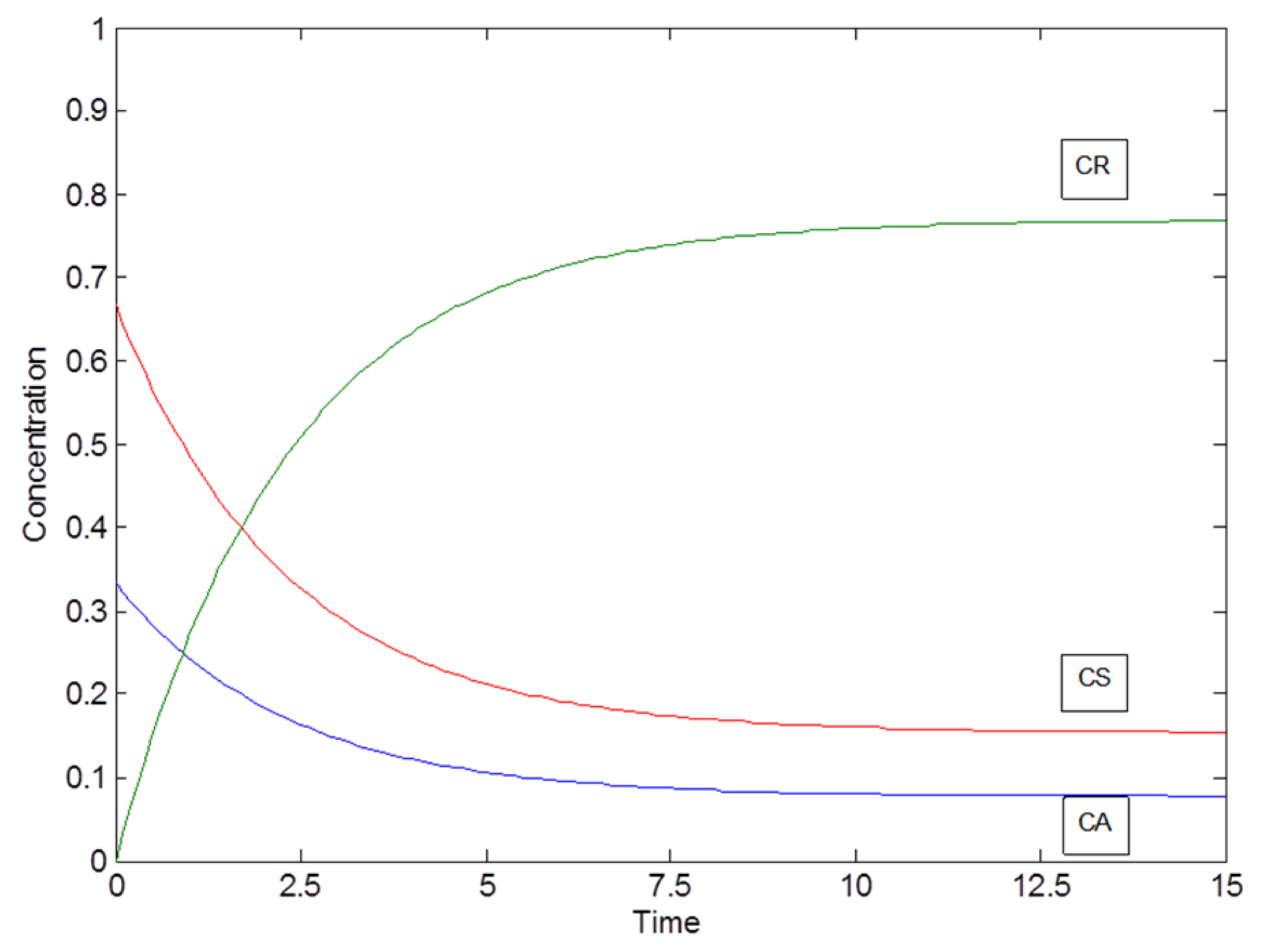
2.1. Example: Consecutive Reactions with Slow and Rapid Steps
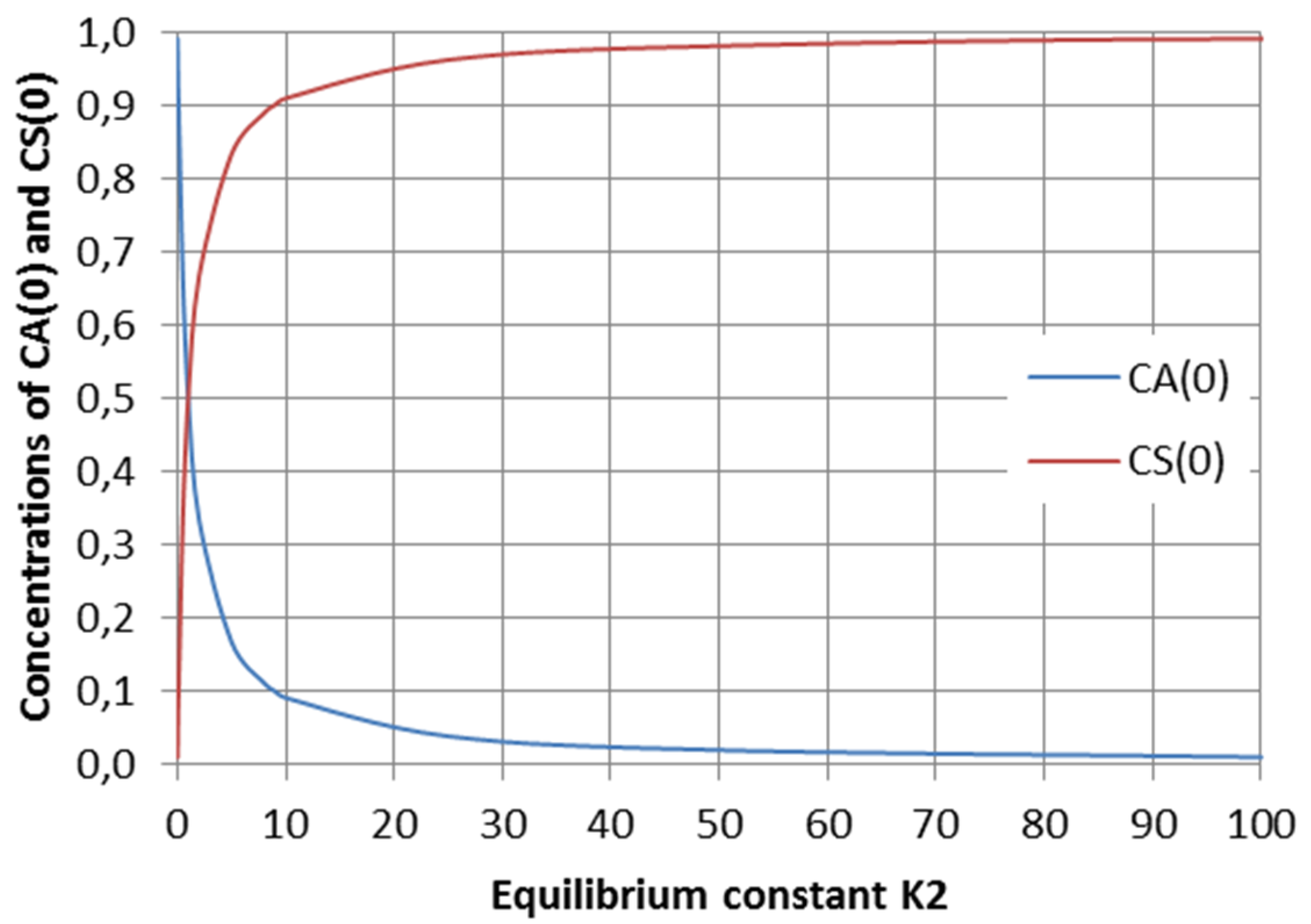
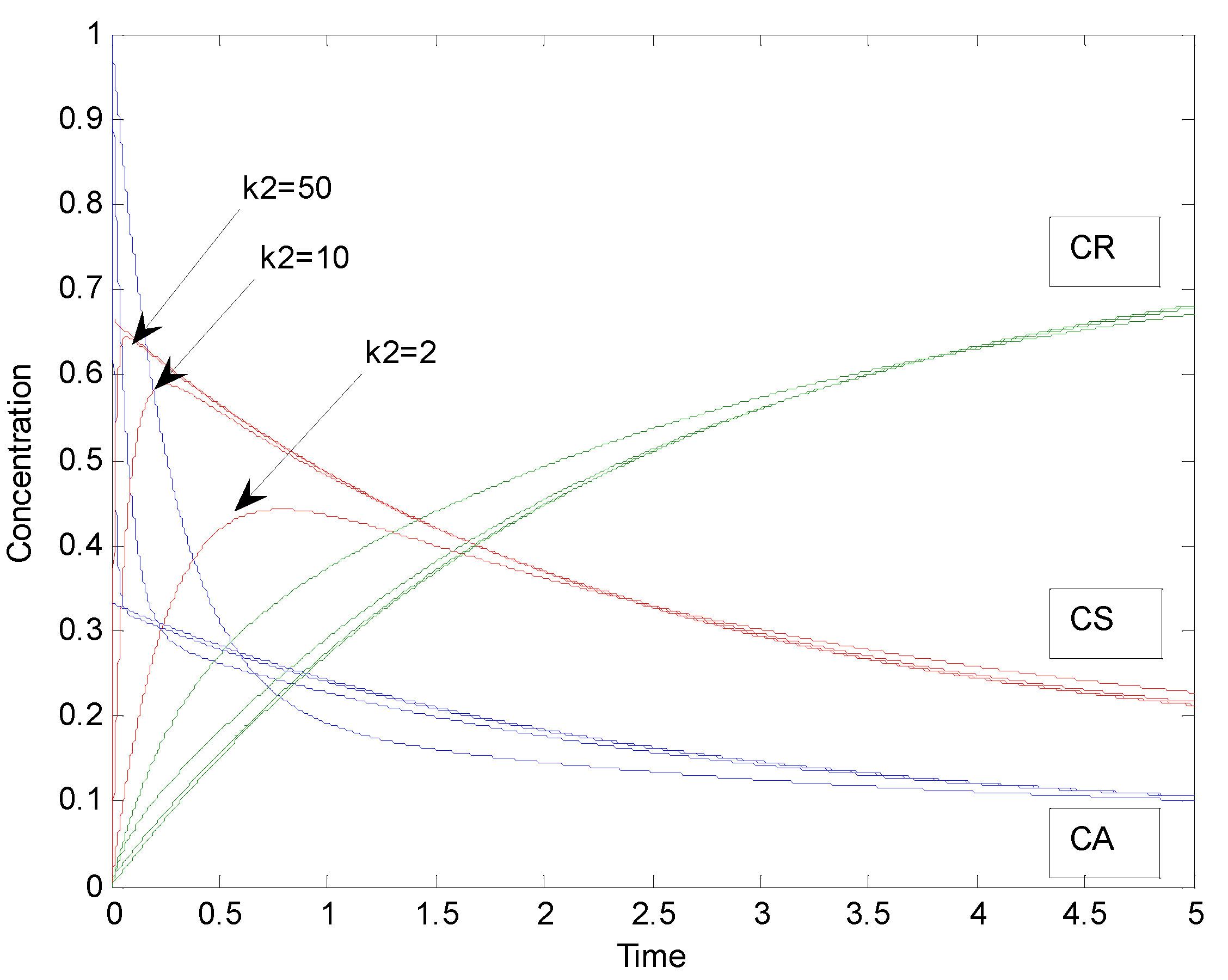
2.2. Parallel Reactions with Slow and Rapid Steps
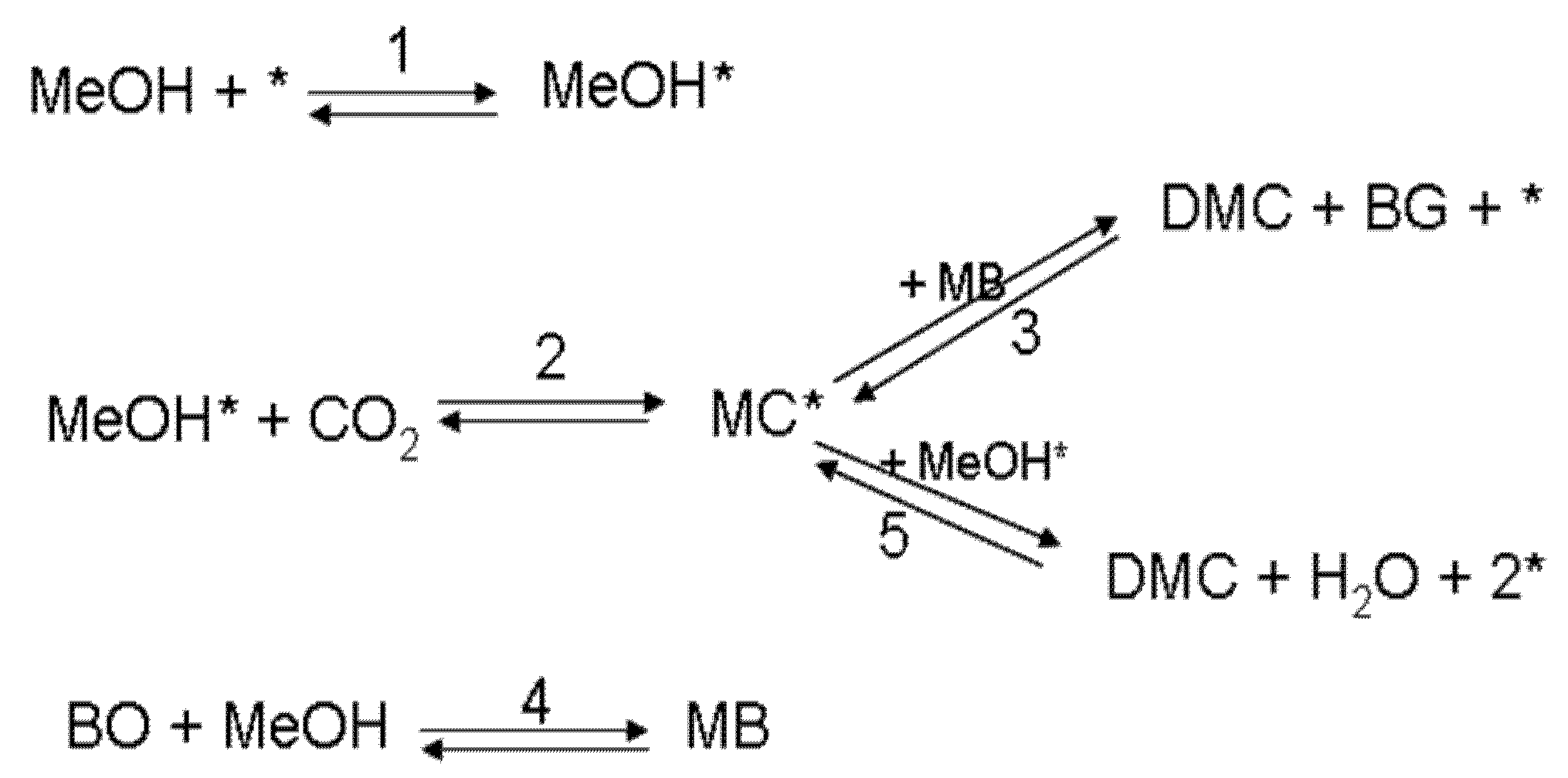
2.3. Example: Synthesis of Dimethyl Carbonate from Methanol and Carbon Dioxide
2.3.1. Basic Mass Balances
2.3.2. Differential-Algebraic Problem
2.3.3. Transformation to ODEs
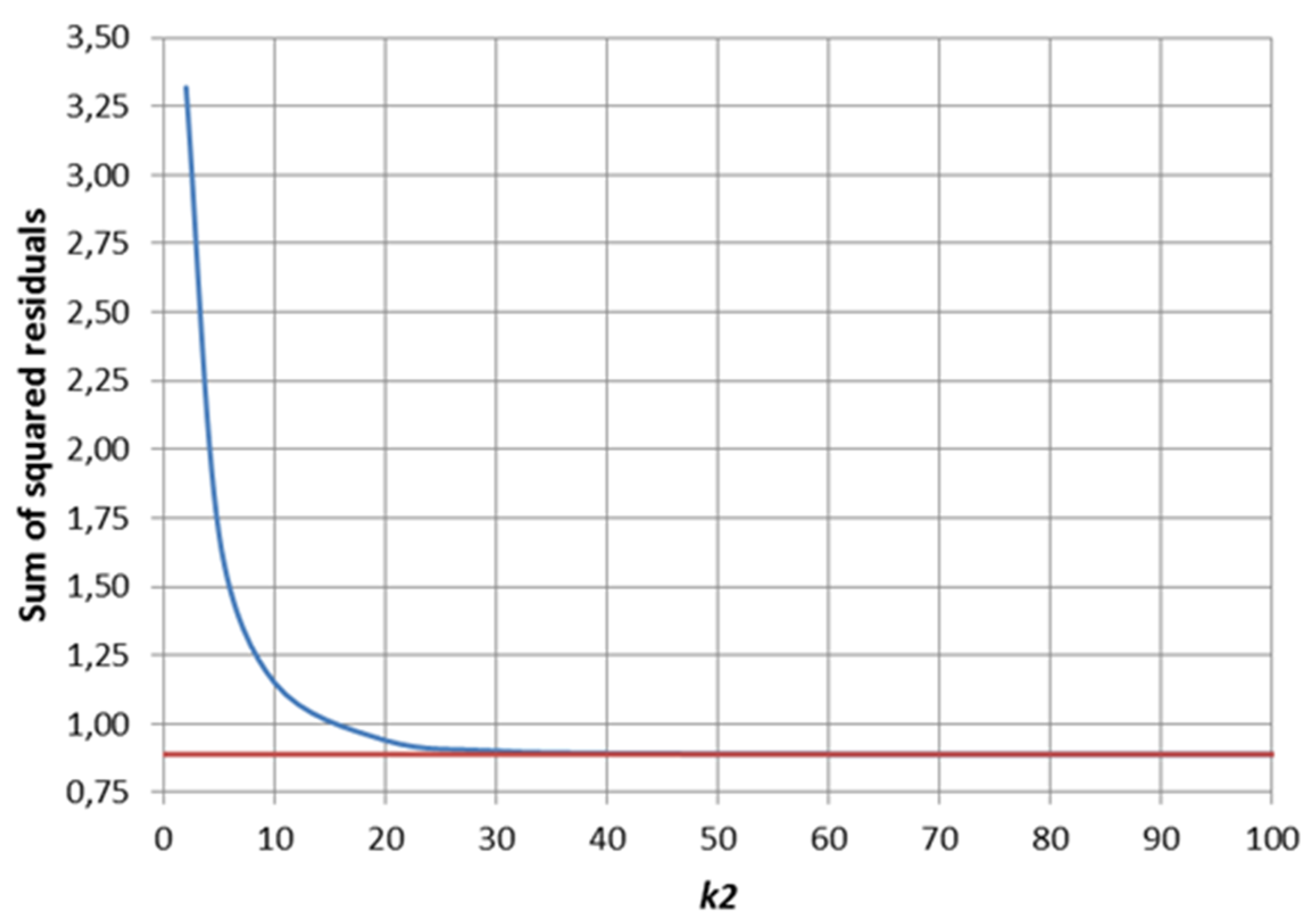
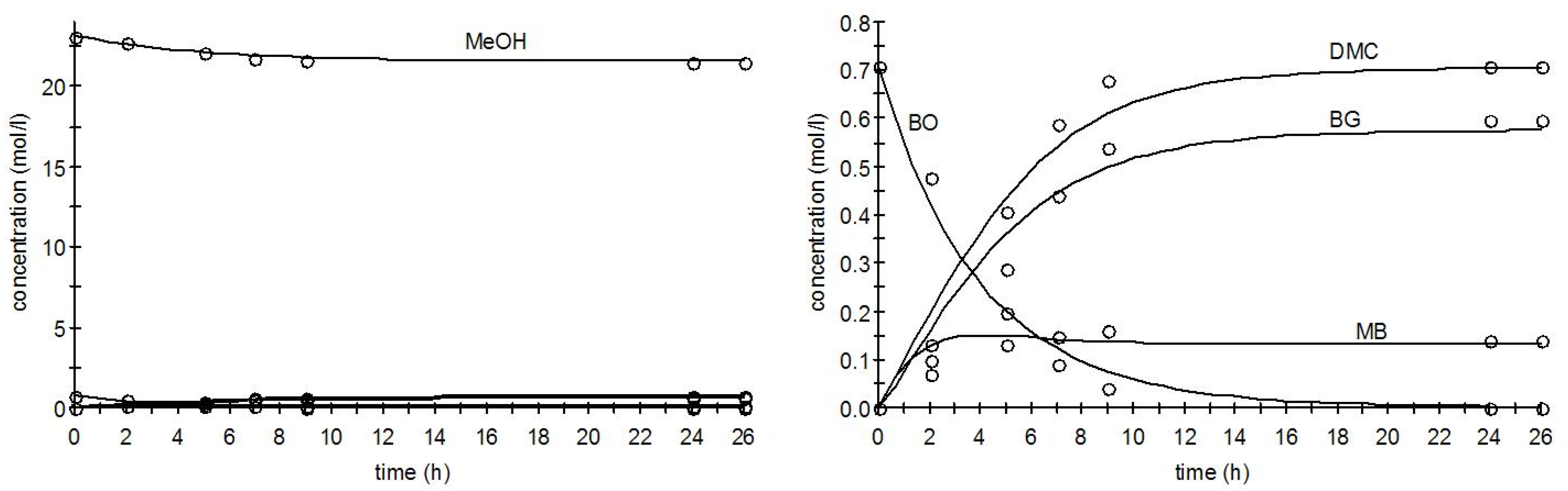
2.3.4. Parameter Estimation Results
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Notation
| c | concentration |
| c* | concentration of an intermediate |
| f | function |
| k | reaction rate constant |
| r | Rate |
| t | Time |
| y | concentration variable |
| α | parameter in rate equation |
| β | parameter in rate equation |
| γ | merged concentration |
| ρB | catalyst bulk density (mass of catalyst-to-liquid volume) |
| ω | merged parameter |
| [] | concentration |
Appendix A. Derivation of the Rate Equations
References
- Jogunola, O.; Salmi, T.; Wärnå, J.; Mikkola, J.-P.; Tirronen, E. Kinetics of methyl formate hydrolysis in the absence and presence of a complexing agent. Ind. Eng. Chem. Res. 2011, 50, 267–276. [Google Scholar] [CrossRef]
- Branco, P.D.; Yablonsky, G.; Marin, G.B.; Constales, D. The switching point between kinetic and thermodynamic control. Comp. Chem. Eng. 2019, 125, 606–611. [Google Scholar] [CrossRef]
- Ono, Y. DMC for environmentally benign reactions. Catal. Today 1997, 35, 15–25. [Google Scholar] [CrossRef]
- Tundo, P. New developments in dimethyl carbonate chemistry. Pure Appl. Chem. 2001, 73, 1117–1124. [Google Scholar] [CrossRef] [Green Version]
- Eta, V.; Mäki-Arvela, P.; Leino, E.; Kordás, K.; Salmi, T.; Murzin, D.; Mikkola, J.-P. Sustainable synthesis of dimethyl carbonate from methanol and carbon dioxide under dehydration- the effect of magnesium enhanced reactions. Ind. Eng. Chem. Res. 2010, 49, 9609–9617. [Google Scholar] [CrossRef]
- Eta, V. Catalytic Synthesis of Dimethyl Carbonate from Carbon Dioxide and Methanol. Ph.D. Thesis, Åbo Akademi, Turku, Finland, 2011. [Google Scholar]
- Haario, H. Modest-User’s Guide; Profmath: Helsinki, Finland, 2007. [Google Scholar]












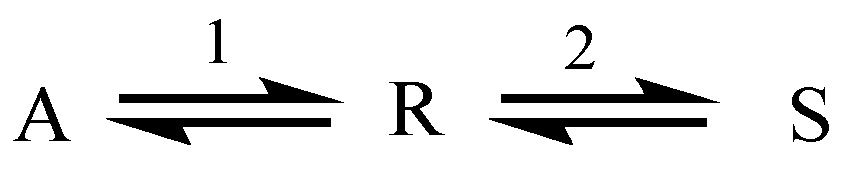{kind=link}
{kind=link}
{kind=link}
{kind=link}
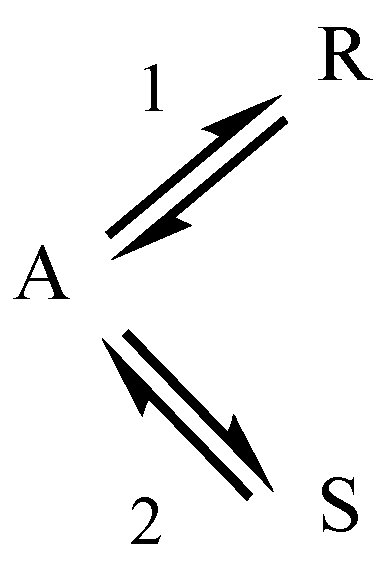{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| k2 | S |
|---|---|
| 1 | 3.6113 |
| 5 | 0.2373 |
| 10 | 0.0644 |
| 100 | 0.006522 |
| 300 | 0.00071645 |
| k2 | S |
|---|---|
| 2 | 3.31901 |
| 5 | 1.68380 |
| 10 | 1.14954 |
| 50 | 0.89085 |
| 100 | 0.88929 |
| 1000 | 0.888893 |
| 10,000 | 0.888889 |
| Parameter | Value | Error/% | |
|---|---|---|---|
| k2 | 1.60 × 10−5 | 6.8 | |
| k4 | 1.12 × 10−2 | 6.7 | |
| K1 = 0, 1/K3 = 0, α = 1.3 × 10−3, K = 0.08 × 10−5, at 150 °C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salmi, T.; Tirronen, E.; Wärnå, J.; Mikkola, J.-P.; Murzin, D.; Eta, V. A Robust Method for the Estimation of Kinetic Parameters for Systems Including Slow and Rapid Reactions—From Differential-Algebraic Model to Differential Model. Processes 2020, 8, 1552. https://doi.org/10.3390/pr8121552
Salmi T, Tirronen E, Wärnå J, Mikkola J-P, Murzin D, Eta V. A Robust Method for the Estimation of Kinetic Parameters for Systems Including Slow and Rapid Reactions—From Differential-Algebraic Model to Differential Model. Processes. 2020; 8(12):1552. https://doi.org/10.3390/pr8121552
Chicago/Turabian StyleSalmi, Tapio, Esko Tirronen, Johan Wärnå, Jyri-Pekka Mikkola, Dmitry Murzin, and Valerie Eta. 2020. "A Robust Method for the Estimation of Kinetic Parameters for Systems Including Slow and Rapid Reactions—From Differential-Algebraic Model to Differential Model" Processes 8, no. 12: 1552. https://doi.org/10.3390/pr8121552
APA StyleSalmi, T., Tirronen, E., Wärnå, J., Mikkola, J. -P., Murzin, D., & Eta, V. (2020). A Robust Method for the Estimation of Kinetic Parameters for Systems Including Slow and Rapid Reactions—From Differential-Algebraic Model to Differential Model. Processes, 8(12), 1552. https://doi.org/10.3390/pr8121552