
Synthesis and Structure of a Ferrocenylsilane-Bridged Bisphosphine †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
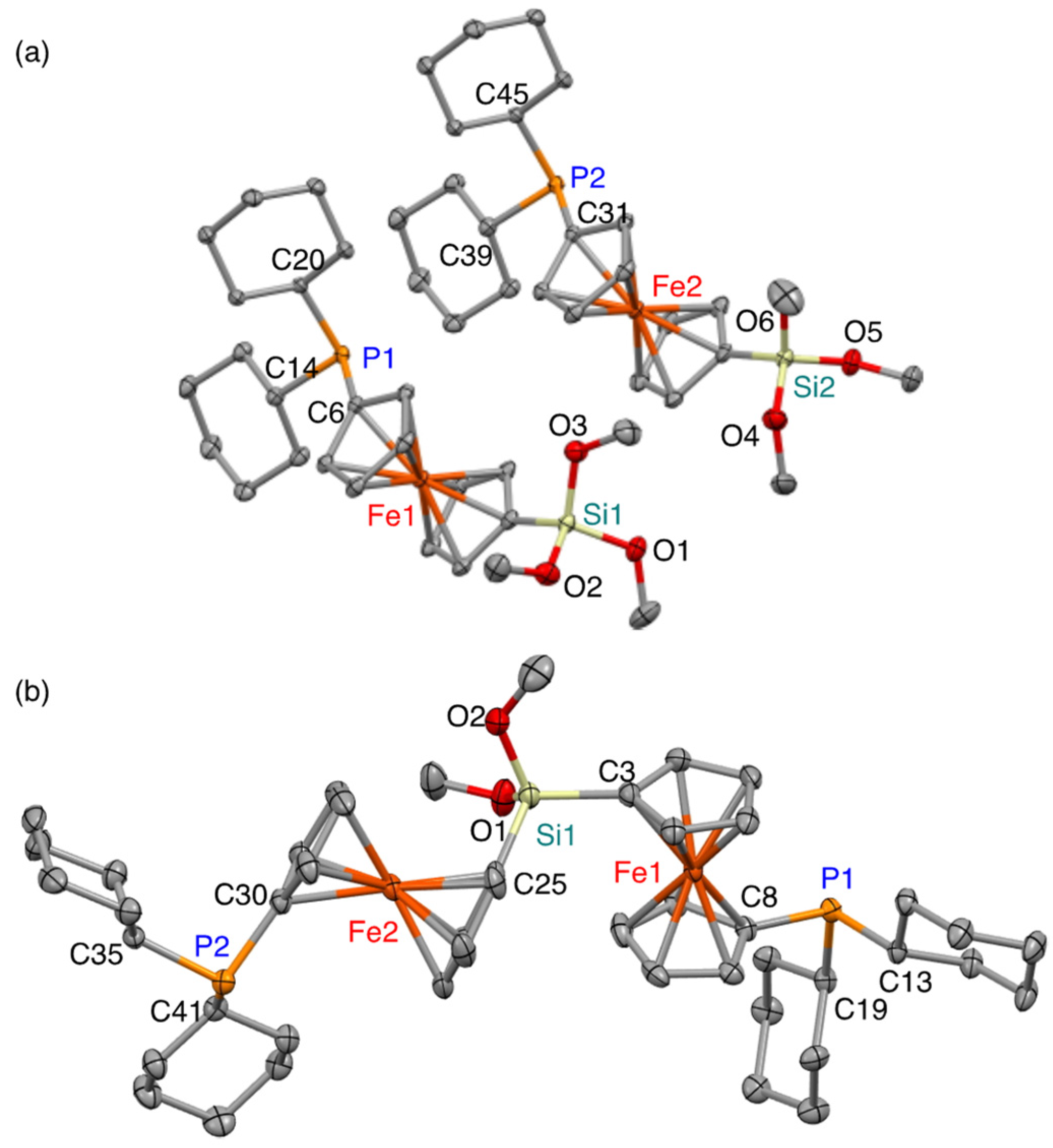
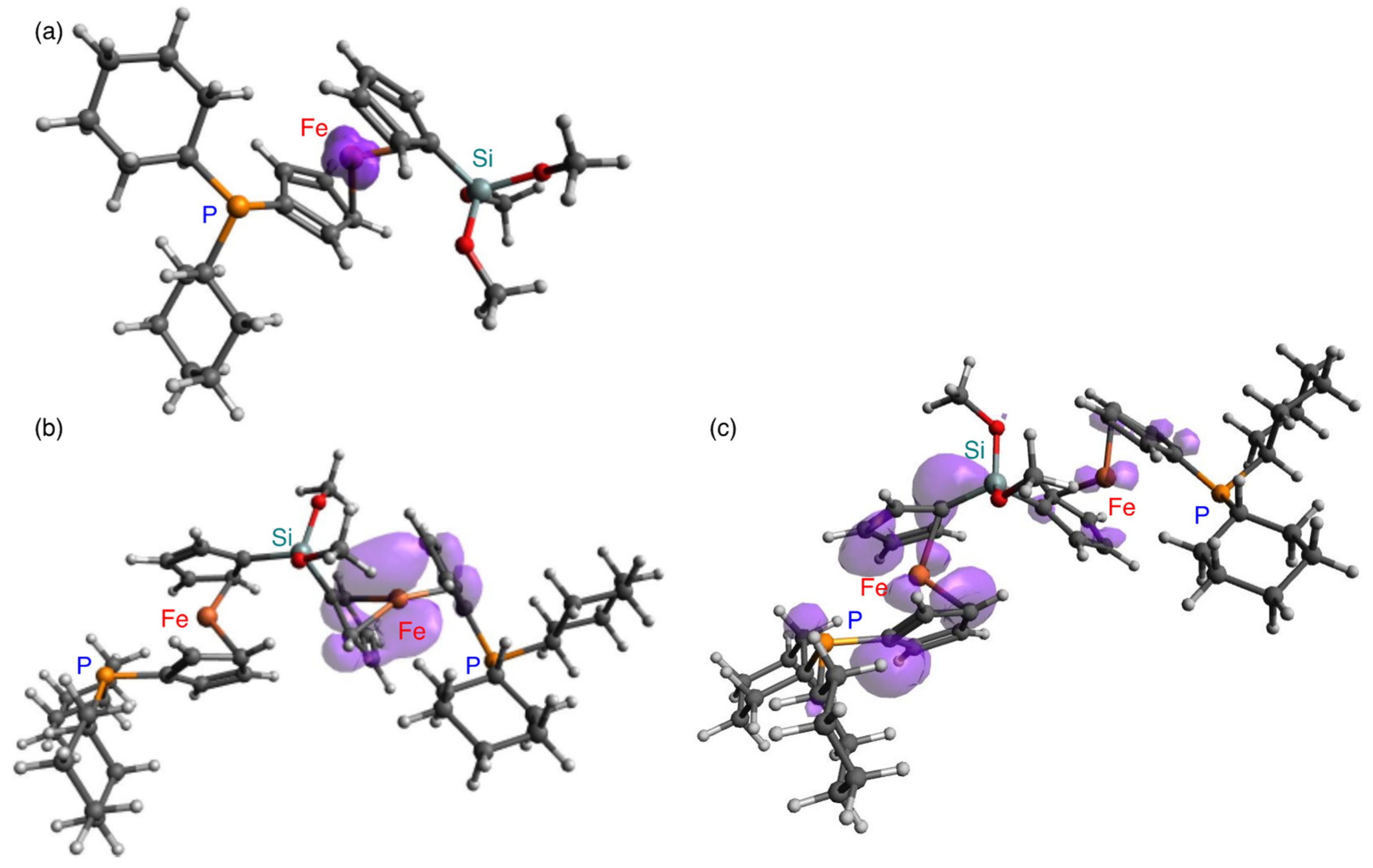
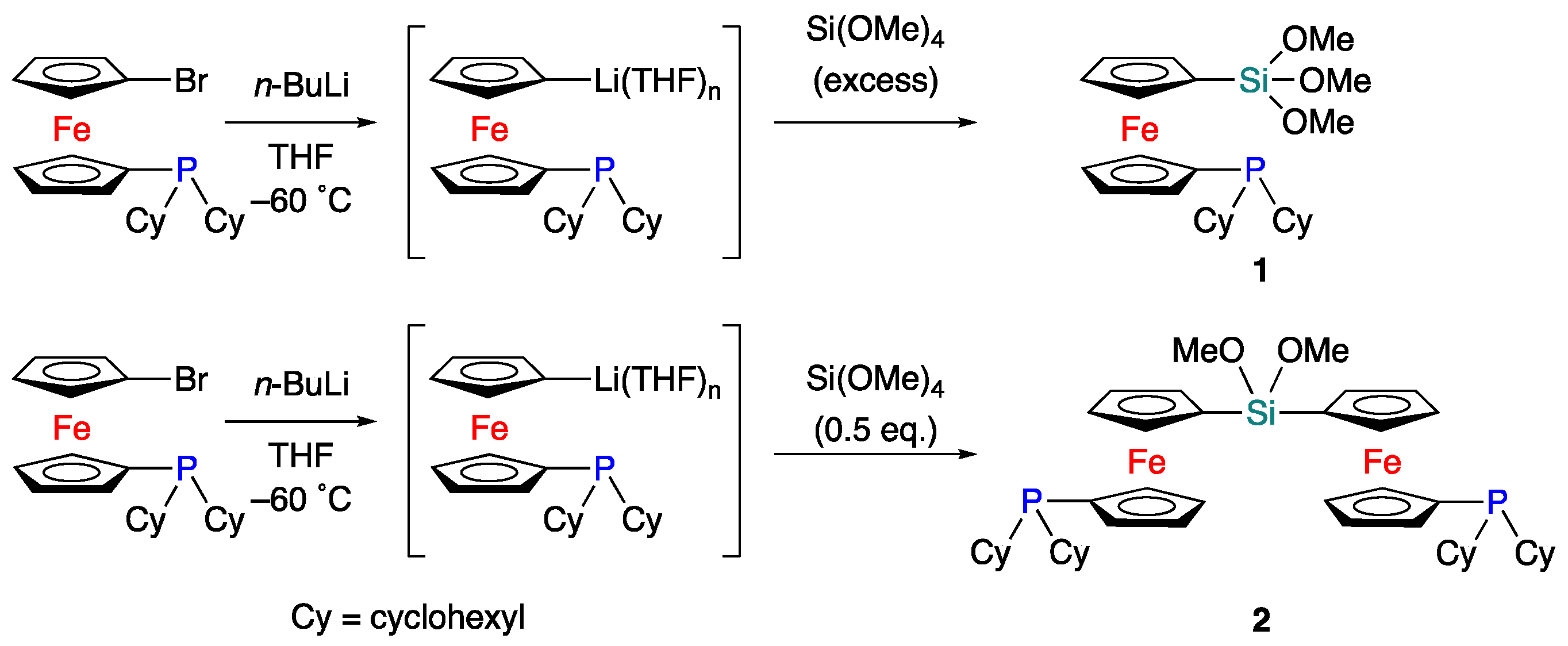
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Synthesis of Monophosphine 1
4.3. Synthesis of Bisphosphine 2
4.4. X-ray Crystallographic Analysis of 1 and 2
4.5. Electrochemical Measurements
4.6. Theoretical Calculations
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamer, P.C.J.; Van Leeuwen, P.W.N.M. Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis; Wiley: Chichester, UK, 2012. [Google Scholar]
- Tang, W.; Zhang, X. New Chiral Phosphorus Ligands for Enantioselective Hydrogenation. Chem. Rev. 2003, 103, 3029–3069. [Google Scholar]
- Shimizu, H.; Nagasaki, I.; Saito, T. Recent advances in biaryl-type bisphosphine ligands. Tetrahedron 2005, 61, 5405–5432. [Google Scholar] [CrossRef]
- Adak, L.; Hatakeyama, T.; Nakamura, M. Iron-Catalyzed Cross-Coupling Reactions Tuned by Bulky Ortho-Phenylene Bisphosphine Ligands. Bull. Chem. Soc. Jpn. 2021, 94, 1125–1141. [Google Scholar] [CrossRef]
- Yu, C.-H.; Yang, X.; Ji, X.; Wang, C.-H.; Lai, Q.; Bhuvanesh, N.; Ozerov, O.V. Redox Communication between Two Diarylamido/Bis(phosphine) (PNP)M Moieties Bridged by Ynediyl Linkers (M = Ni, Pd, Pt). Inorg. Chem. 2020, 59, 10153–10162. [Google Scholar] [CrossRef] [PubMed]
- Orton, G.R.F.; Pilgrim, B.S.; Champness, N.R. The chemistry of phosphines in constrained, well-defined microenvironments. Chem. Soc. Rev. 2021, 50, 4411–4431. [Google Scholar] [CrossRef] [PubMed]
- Bezrukov, A.A.; Törnroos, K.W.; Dietzel, P.D.C. Modification of Network and Pore Dimensionality in Metal−Organic Frameworks Containing a Secondary Phosphine Functionality. Cryst. Growth Des. 2017, 17, 3257–3266. [Google Scholar] [CrossRef]
- Falkowski, J.M.; Sawano, T.; Zhang, T.; Tsun, G.; Chen, Y.; Lockard, J.V.; Lin, W. Privileged Phosphine-Based Metal−Organic Frameworks for Broad-Scope Asymmetric Catalysis. J. Am. Chem. Soc. 2014, 136, 5213–5216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lou, L.-L.; Yu, K.; Liu, S. Advances in Chiral Metal–Organic and Covalent Organic Frameworks for Asymmetric Catalysis. Small 2021, 17, 2005686. [Google Scholar] [CrossRef] [PubMed]
- Štěpnička, P. Ferrocenes: Ligands, Materials and Biomolecules; John Wiley & Sons Ltd.: Chichester, UK, 2008. [Google Scholar] [CrossRef]
- Togni, A.; Hayashi, T. Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science; VCH: New York, NY, USA, 1995. [Google Scholar]
- Ringenberg, M.R.; Wittkamp, F.; Apfel, U.P.; Kaim, W. Redox Induced Configurational Isomerization of Bisphosphine-Tricarbonyliron(I) Complexes and the Difference a Ferrocene Makes. Inorg. Chem. 2017, 56, 7501–7511. [Google Scholar] [CrossRef] [PubMed]
- Loschen, R.; Loschen, C.; Frank, W.; Ganter, C. Synthesis, Structure and Reactivity of Trimethylsilyl-Substituted Phosphametallocenes. Eur. J. Inorg. Chem. 2007, 2007, 553–561. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.; Yun, J.; Kong, J. Novel ferrocene-containing organosilicon polymers and uniform microspheres prepared by free radical copolymerization: Precursors for magnetic Si-C-Fe-(O) nanomaterials. Mater. Des. 2018, 144, 86–97. [Google Scholar] [CrossRef]
- Manners, I. Ring-opening polymerization of strained metallocenophanes: A new route to high molecular weight poly(metallocenes). Pure Appl. Chem. 1999, 71, 1471–1476. [Google Scholar] [CrossRef]
- Herbert, D.E.; Mayer, U.F.J.; Manners, I. Strained Metallocenophanes and Related Organometallic Rings Containing π-Hydrocarbon Ligands and Transition-Metal Centers. Angew. Chem. Ind. Ed. 2007, 46, 5060–5081. [Google Scholar] [CrossRef] [PubMed]
- Bruña, S.; Martínez-Montero, I.; González-Vadillo, A.M.; Martín-Fernández, C.; Montero-Campillo, M.M.; Mó, O.; Cuadrado, I. Ferrocene and Silicon-Containing Oxathiacrown Macrocycles and Linear Oligo-Oxathioethers Obtained via Thiol−Ene Chemistry of a Redox-Active Bifunctional Vinyldisiloxane. Macromolecules 2015, 48, 6955–6969. [Google Scholar] [CrossRef]
- Ma, Q.; Qi, Y.; Li, J.; Wang, W.; Sun, X. A ferrocene-containing porous organic polymer linked by tetrahedral silicon-centered units for gas sorption. Appl. Organometal. Chem. 2018, 32, e3935. [Google Scholar] [CrossRef]
- Hussein, M.A.; Asiri, A.M. Organometallic Ferrocene- and Phosphorus-Containing Polymers: Synthesis and Characterization. Des. Monomers Polym. 2012, 15, 207–251. [Google Scholar] [CrossRef]
- Štěpnička, P.; Císařová, I. Selective borane reduction of phosphinoferrocene carbaldehydes to phosphinoalcohol-borane adducts. The coordination behaviour of 1-(diphenylphosphino)-1 ’-(methoxymethyl)ferrocene, a new ferrocene O,P-hybrid donor prepared from such an adduct. Dalton Trans. 2013, 42, 3373–3389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lee, V.Y.; Morisako, S.; Aoyagi, S.; Sasamori, T. Ferrocene-Based Phosphenium Ion with Intramolecular Phosphine Coordination. Eur. J. Inorg. Chem. 2021, 2021, 3988–3991. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Richardson, D.E.; Taube, H. Mixed-valence molecules: Electronic delocalization and stabilization. Coord. Chem. Rev. 1984, 60, 107–129. [Google Scholar] [CrossRef]
- Usón, I.; Sheldrick, G.M. An introduction to experimental phasing of macromolecules illustrated by SHELX; new autotracing features. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sasamori, T.; Ueno, H.; Morisako, S. Synthesis and Structure of a Ferrocenylsilane-Bridged Bisphosphine. Inorganics 2022, 10, 22. https://doi.org/10.3390/inorganics10020022
Sasamori T, Ueno H, Morisako S. Synthesis and Structure of a Ferrocenylsilane-Bridged Bisphosphine. Inorganics. 2022; 10(2):22. https://doi.org/10.3390/inorganics10020022
Chicago/Turabian StyleSasamori, Takahiro, Hiromu Ueno, and Shogo Morisako. 2022. "Synthesis and Structure of a Ferrocenylsilane-Bridged Bisphosphine" Inorganics 10, no. 2: 22. https://doi.org/10.3390/inorganics10020022
APA StyleSasamori, T., Ueno, H., & Morisako, S. (2022). Synthesis and Structure of a Ferrocenylsilane-Bridged Bisphosphine. Inorganics, 10(2), 22. https://doi.org/10.3390/inorganics10020022