

Ferrocene-Bearing Homoleptic and Heteroleptic Paddlewheel-Type Dirhodium Complexes



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization
2.2. Crystal Structures
2.3. Optimized Geometries and Electronic Structures
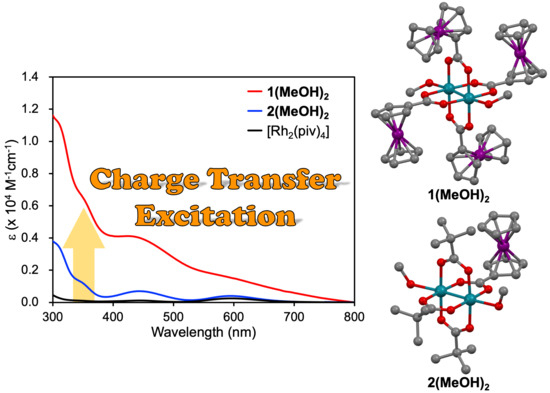
2.4. Absorption Features
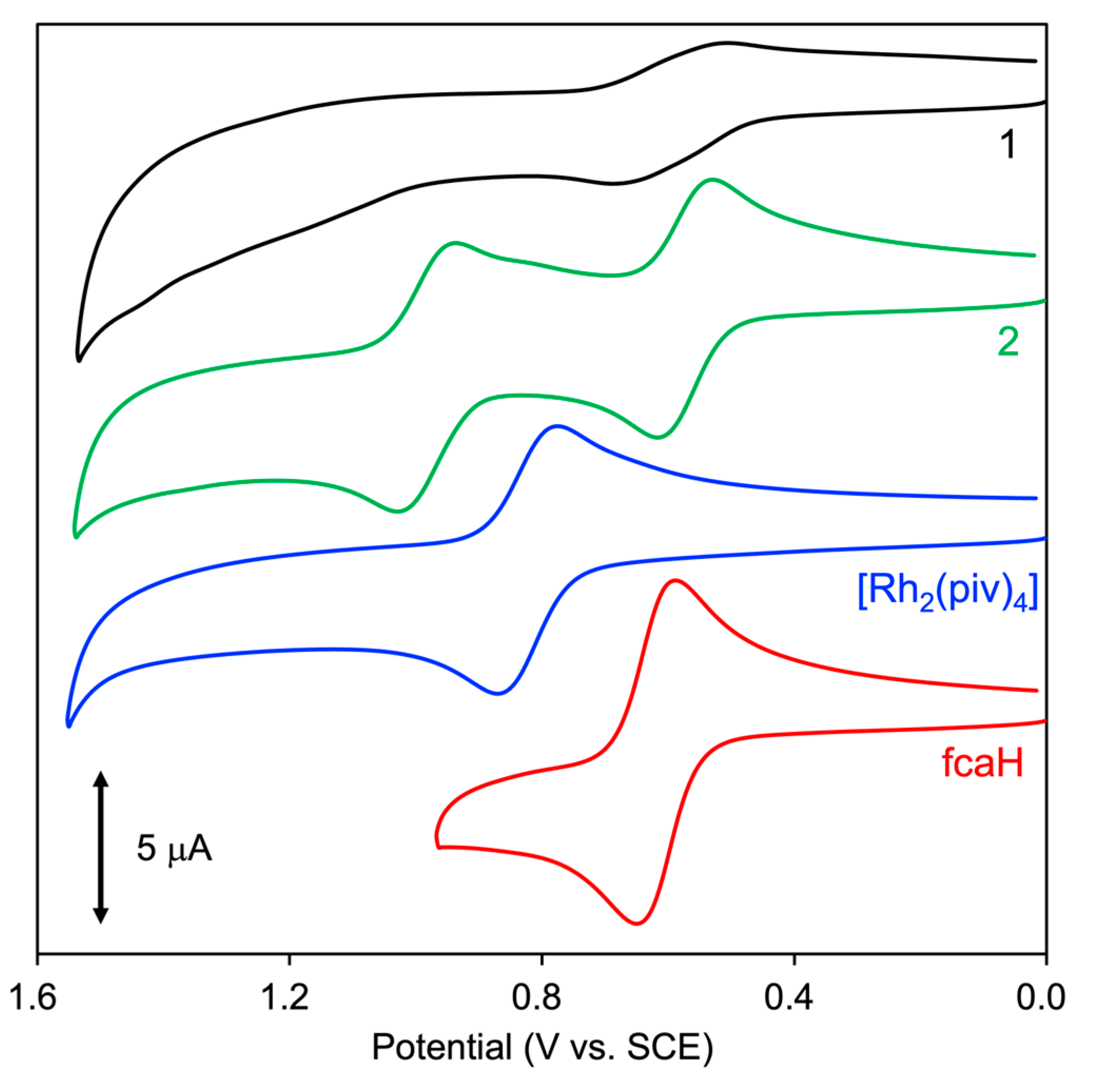
2.5. Electrochemical Properties
3. Materials and Methods
3.1. Chemicals and Instruments
3.2. Synthesis of [Rh2(fca)4] (1)
3.3. Synthesis of [Rh2(fca)(piv)3] (2)
3.4. Crystallography
3.5. Theoretical Calculation Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jansze, S.M.; Severin, K. Clathrochelate Metalloligands in Supramolecular Chemistry and Materials Science. Acc. Chem. Res. 2018, 51, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, N.; Kuwamura, N.; Kojima, T.; Konno, T. Development of coordination chemistry with thiol-containing amino acids. Coord. Chem. Rev. 2023, 474, 214857. [Google Scholar] [CrossRef]
- Kumar, G.; Gupta, R. Molecularly designed architectures—The metalloligand way. Chem. Soc. Rev. 2013, 42, 9403–9453. [Google Scholar] [CrossRef] [PubMed]
- Garibay, S.J.; Stork, J.R.; Cohen, S.M. The Use of Metalloligands in Metal-Organic Frameworks. Prod. Inorg. Chem. 2009, 56, 335–378. [Google Scholar]
- Das, M.C.; Xiang, S.; Zhang, Z.; Chen, B. Functional Mixed Metal-Organic Frameworks with Metalloligands. Angew. Chem. Int. Ed. 2011, 50, 10510–10520. [Google Scholar] [CrossRef]
- Gao, W.X.; Zhang, H.N.; Jin, G.X. Supramolecular catalysis based on discrete heterometallic coordination-driven metallacycles and metallacages. Coord. Chem. Rev. 2019, 386, 69–84. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Takeda, H.; Ishitani, O. Photocatalytic reduction of CO2 using metal complexes. J. Photochem. Photobiol. C-Photochem. Rev. 2015, 25, 106–137. [Google Scholar] [CrossRef]
- Rau, S.; Schäfer, B.; Gleich, D.; Anders, E.; Rudolph, M.; Friedrich, M.; Görls, H.; Henry, W.; Vos, J.G. A supramolecular photocatalyst for the production of hydrogen and the selective hydrogenation of tolane. Angew. Chem. Int. Ed. 2006, 45, 6215–6218. [Google Scholar] [CrossRef]
- Singh, A.K.; Pandey, D.S.; Xu, Q.; Braunstein, P. Recent advances in supramolecular and biological aspects of arene ruthenium(II) complexes. Coord. Chem. Rev. 2014, 270, 31–56. [Google Scholar] [CrossRef]
- Gil-Rubio, J.; Vicente, J. The Coordination and Supramolecular Chemistry of Gold Metalloligands. Chem. Eur. J. 2018, 24, 32–46. [Google Scholar] [CrossRef]
- Zheng, X.Y.; Kong, X.J.; Zheng, Z.; Long, L.S.; Zheng, L.S. High-Nuclearity Lanthanide-Containing Clusters as Potential Molecular Magnetic Coolers. Acc. Chem. Res. 2018, 51, 517–525. [Google Scholar] [CrossRef]
- Chiu, C.C.; Cheng, M.C.; Lin, S.H.; Yan, C.W.; Lee, G.H.; Chang, M.C.; Lin, T.S.; Peng, S.M. Structure and magnetic properties of a novel heteroheptanuclear metal string complex [Ni3Ru2Ni2(μ7-teptra)4(NCS)2](PF6). Dalton Trans. 2020, 49, 6643–6653. [Google Scholar] [CrossRef]
- Cotton, F.A.; Reid, A.H. Solid-State structure of Ferrocenecarboxylic acid, [Fe(C5H4CO2H)(C5H5)]. Acta Cryst. 1985, C41, 686–688. [Google Scholar] [CrossRef]
- Prokopuk, N.; Shriver, D.F. A one-dimensional array of clusters: Na2Mo6Cl8(O2CC5H4FeCp)6·CH3OH. Inorg. Chem. 1997, 36, 5609–5613. [Google Scholar] [CrossRef]
- Fan, Y.; Cui, Y.; Zou, G.D.; Duan, R.H.; Zhang, X.; Dong, Y.X.; Lv, H.T.; Cao, J.T.; Jing, Q.S. A ferrocenecarboxylate-functionalized titanium-oxo-cluster: The ferrocene wheel as a sensitizer for photocurrent response. Dalton Trans. 2017, 46, 8057–8064. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Li, G.; Li, L.; Zhu, Y.; Meng, X.; Fan, Y. Synthesis, crystal structures, and magnetic properties of three novel ferrocenecarboxylato-bridged lanthanide dimers. Inorg. Chem. 2003, 42, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Mashima, K. Interaction of Ferrocene Moieties Across a Square Pt4 Unit: Synthesis, Characterization, and Electrochemical Properties of Carboxylate-Bridged Bimetallic Pt4Fen (n = 2, 3, and 4) Complexes. Inorg. Chem. 2011, 50, 11384–11393. [Google Scholar] [CrossRef]
- Cotton, F.A.; Murillo, C.A.; Walton, R.A. Multiple Bonds between Metal Atoms, 3rd ed.; Springer Science and Business Media: New York, NY, USA, 2005. [Google Scholar]
- Churchill, M.R.; Li, Y.J.; Nalewajek, D.; Schaber, P.M.; Dorfman, J. Preparation, Crystal and Molecular Structure, and Properties of Tetrakis(ferrocenecarboxylato)bis(tetrahydrofuran)dicopper(II). A Structure Containing both Eclipsed and Staggered Ferrocenyl Fragments. Inorg. Chem. 1985, 24, 2684–2687. [Google Scholar] [CrossRef]
- Artetxe, B.; Vitoria, P.; Pache, A.; Reinoso, S.; Gutiérrez-Zorrilla, J.M. Tetrakis(μ2-ferrocenecarboxylato-κ2O:O′)bis[(methanol-κO)copper(II)] methanol disolvate. Acta Cryst. 2011, E67, m1840–m1841. [Google Scholar] [CrossRef]
- Zhang, E.; Hou, H.; Meng, X.; Liu, Y.; Liu, Y.; Fan, Y. Ferrocenyl Functional Coordination Polymers Based on Mono-, Bi-, and Heterotrinuclear Organometallic Building Blocks: Syntheses, Structures, and Properties. Cryst. Growth Des. 2009, 9, 903–913. [Google Scholar] [CrossRef]
- Cotton, F.A.; Falvello, L.R.; Reid, A.H.; Tocher, J.H. Mixed-ligand systems containing quadruple bonds. Capture and structural characterization of an intermediate in the ligand exchange process leading to new carboxylates of the dimolybdenum(4+) unit. Synthesis and X-ray crystallographic and electrochemical studies of Mo2[(η5-C5H4CO2)Fe(η5-C5H5)]2(O2CCH3)2(C5H5N)2 and [Mo2](η5-C5H4CO2)Fe(η5-C5H5)]4(ax-CH3CN)(ax-DMSO)](DMSO)2. J. Organomet. Chem. 1987, 319, 87–97. [Google Scholar]
- Cai, X.M.; Meister, T.K.; Pöthig, A.; Kühn, F.E. Filling a Gap: Electrochemical Property Comparison of the Completed Compound Series [Mo2(DArF)n(O2C-Fc)4–n] (DArF = N,N′-Diarylformamidinate; O2C-Fc = Ferrocenecarboxylate). Inorg. Chem. 2016, 55, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.W.; Cameron, T.S.; Robertson, K.N.; Swarts, J.C.; Aquino, M.A.S. Structure and electrochemistry of various diruthenium(II,III) tetrametallocenecarboxylates. Organometallics 2002, 21, 5962–5971. [Google Scholar] [CrossRef]
- Boyd, D.A.; Cao, Z.; Song, Y.; Wang, T.W.; Fanwick, P.E.; Crutchley, R.J.; Ren, T. Diruthenium Compounds Bearing Equatorial Fc-containing Ligands: Synthesis and Electronic Structure. Inorg. Chem. 2010, 49, 11525–11531. [Google Scholar] [CrossRef] [PubMed]
- Noels, A.F.; Demonceau, A.; Petiniot, N.; Hubert, A.J.; Teyssie, P. Transition-metal-catalyzed reaction of diazocompounds, efficient synthesis of functionalized ethers by carbene insertion into the hydroxylic bond of alcohols. Tetrahedron 1982, 38, 2733–2739. [Google Scholar] [CrossRef]
- Demonceau, A.; Noels, A.F.; Teyssie, P.; Hubert, A.J. Shape selective alkane functionalisation by ethyl diazoacetate catalysed by rhodium carboxylates. J. Mol. Catal. 1988, 49, L13–L17. [Google Scholar] [CrossRef]
- Vosáhlo, P.; Harmach, P.; Císarová, I.; Stepnicka, P. Synthesis and characterisation of dirhodium(II) tetraacetates bearing axial ferrocene ligands. J. Organomet. Chem. 2021, 953, 122065. [Google Scholar] [CrossRef]
- Hansen, J.; Davies, H.M.L. High Symmetry Dirhodium(II) Paddlewheel Complexes as Chiral Catalysts. Coord. Chem. Rev. 2008, 252, 545–555. [Google Scholar] [CrossRef]
- Fiori, K.W.; Du Bois, J. Catalytic Intermolecular Amination of C−H Bonds: Method Development and Mechanistic Insights. J. Am. Chem. Soc. 2007, 129, 562–568. [Google Scholar] [CrossRef]
- Kataoka, Y.; Yano, N.; Handa, M.; Kawamoto, T. Intrinsic Hydrogen Evolution Capability and Theoretically Supported Reaction Mechanism of Paddlewheel-type Dirhodium Complex. Dalton Trans. 2019, 48, 7302–7312. [Google Scholar] [CrossRef]
- Kataoka, Y.; Sato, K.; Yano, N. Hydroxypyridinate-bridged paddlewheel-type dirhodium complex as a catalyst for photochemical and electrochemical hydrogen evolution. J. Chem. Phys. 2023, 159, 204304. [Google Scholar] [CrossRef] [PubMed]
- Hilderbrand, S.A.; Lim, M.H.; Lippard, S.J. Dirhodium Tetracarboxylate Scaffolds as Reversible Fluorescence-Based Nitric Oxide Sensors. J. Am. Chem. Soc. 2004, 126, 4972–4978. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, Y.; Kohara, Y.; Yano, N.; Kawamoto, T. Unique vapochromism of a paddlewheel-type dirhodium complex accompanied by dynamic structural and phase transitions. Dalton Trans. 2020, 49, 14373–14377. [Google Scholar] [CrossRef]
- Chifotides, H.T.; Dunber, K.R. Interactions of Metal−Metal-Bonded Antitumor Active Complexes with DNA Fragments and DNA. Acc. Chem. Res. 2005, 38, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, Y.; Yano, N.; Mikuriya, M.; Handa, M. Coordination polymers and metal–organic frameworks based on paddlewheel-type dirhodium(II) tetracarboxylates. Coord. Chem. Rev. 2022, 472, 214796. [Google Scholar] [CrossRef]
- Kataoka, Y.; Yano, N.; Mikuriya, M.; Handa, M. Paddlewheel-type dirhodium complexes with N,N’-bridging ligands. Coord. Chem. Rev. 2023, 479, 214997. [Google Scholar] [CrossRef]
- Sawamura, M.; Sasaki, H.; Nakata, T.; Ito, Y. Synthesis of Optically Active Ferrocene Analogues of Salicylic Acid Derivatives and Rhodium(II)-Catalyzed Asymmetric Intramolecular C–H Insertion of α-Diazo β-Keto Esters Using New Chiral Carboxylato Ligands. Bull. Chem. Soc. Jpn. 1993, 66, 2725–2729. [Google Scholar] [CrossRef]
- Arakawa, K.; Yano, N.; Imasaki, N.; Kohara, Y.; Yatsushiro, D.; Atarashi, D.; Handa, M.; Kataoka, Y. Coordination-Induced Self-Assembly of a Heteroleptic Paddlewheel-Type Dirhodium Complex. Crystals 2020, 10, 85. [Google Scholar] [CrossRef]
- Cotton, F.A.; Felthouse, T.R. Structural studies of three tetrakis(carboxylato)dirhodium(II) adducts in which carboxylate groups and axial ligands are varied. Inorg. Chem. 1980, 19, 323–328. [Google Scholar] [CrossRef]
- Kataoka, Y.; Fukumoto, R.; Yano, N.; Atarashi, D.; Tanaka, H.; Kawamoto, T.; Handa, M. Synthesis, Characterization, Absorption Properties, and Electronic Structures of Paddlewheel-Type Dirhodium(II) Tetra-μ-(n-naphthoate) Complexes: An Experimental and Theoretical Study. Molecules 2019, 24, 447. [Google Scholar] [CrossRef]
- Sizova, O.V.; Skripnikov, L.V.; Sokolov, A.Y.; Ivanova, N.V. Rhodium and ruthenium tetracarboxylate nitrosyl complexes: Electronic structure and metal-metal bond. Russ. J. Inorg. Chem. 2007, 33, 588–593. [Google Scholar] [CrossRef]
- Norman, J.G.; Kolari, H.J. Strength and trans influence of Rh-Rh bond in rhodium(II) carboxylate dimers. J. Am. Chem. Soc. 1978, 100, 791–799. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, H.; Sun, M.; Liu, Y.; Pang, X.; Yu, X.; Liu, B.; Li, Z. Substitution effect on the geometry and electronic structure of the ferrocene. J. Comput. Chem. 2007, 28, 2260–2274. [Google Scholar] [CrossRef] [PubMed]
- Legzdins, P.; Mitchell, R.W.; Rempel, G.L.; Ruddick, J.D.; Wilkinsin, G. The protonation of ruthenium- and rhodium-bridged carboxylates and their use as homogeneous hydrogenation catalysts for unsaturated substances. J. Chem. Soc. A 1970, 3322–3326. [Google Scholar] [CrossRef]
- Kataoka, Y.; Yano, N.; Kawamoto, T.; Handa, M. Isolation of the Intermediate in the Synthesis of Paddlewheel-type Dirhodium Tetraacetate. Eur. J. Inorg. Chem. 2015, 34, 5650–5655. [Google Scholar] [CrossRef]
- CrysAlisPro Software System; Rigaku Oxford Diffraction: Tokyo, Japan, 2018.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- John, D. Roy, Keith, Todd, Millam, GaussView 5.0; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1(MeOH)2 | 2(MeOH)2 | |
|---|---|---|
| Chemical formula | C24H26Fe2O6Rh | C56H88Fe2O20Rh4 |
| Formula weight | 625.06 | 1604.60 |
| Crystal system | Monoclinic | Triclinic |
| Space group | P 21/n | P-1 |
| a (Å) | 13.8360(3) | 13.8890(3) |
| b (Å) | 9.5473(2) | 14.2283(3) |
| c (Å) | 17.6017(4) | 18.1715(4) |
| α (deg) | 90 | 105.982(2) |
| β (deg) | 104.691(2) | 91.795(2) |
| γ (deg) | 90 | 103.012(2) |
| V (Å3) | 2249.11(9) | 3346.82(13) |
| Z | 4 | 2 |
| Dcalc (g cm−3) | 1.846 | 1.592 |
| μ (mm−1) | 2.038 | 1.451 |
| F (000) | 1260.0 | 1632.0 |
| R1 (I > 2σ(I)) | 0.0248 | 0.0403 |
| wR2 (I > 2σ(I)) | 0.0559 | 0.1076 |
| R1 (all data) | 0.0305 | 0.0512 |
| wR2 (all data) | 0.0582 | 0.1138 |
| GOF on F2 | 1.064 | 1.071 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kataoka, Y.; Sato, K.; Yano, N.; Handa, M. Ferrocene-Bearing Homoleptic and Heteroleptic Paddlewheel-Type Dirhodium Complexes. Inorganics 2024, 12, 41. https://doi.org/10.3390/inorganics12020041
Kataoka Y, Sato K, Yano N, Handa M. Ferrocene-Bearing Homoleptic and Heteroleptic Paddlewheel-Type Dirhodium Complexes. Inorganics. 2024; 12(2):41. https://doi.org/10.3390/inorganics12020041
Chicago/Turabian StyleKataoka, Yusuke, Kozo Sato, Natsumi Yano, and Makoto Handa. 2024. "Ferrocene-Bearing Homoleptic and Heteroleptic Paddlewheel-Type Dirhodium Complexes" Inorganics 12, no. 2: 41. https://doi.org/10.3390/inorganics12020041
APA StyleKataoka, Y., Sato, K., Yano, N., & Handa, M. (2024). Ferrocene-Bearing Homoleptic and Heteroleptic Paddlewheel-Type Dirhodium Complexes. Inorganics, 12(2), 41. https://doi.org/10.3390/inorganics12020041