
A Core and Valence-Level Spectroscopy Study of the Enhanced Reduction of CeO2 by Iron Substitution—Implications for the Thermal Water-Splitting Reaction


Abstract
:1. Introduction
- Size substitution (iso-valency): Compensation for lattice expansion. Upon the reduction of Ce4+ cations to Ce3+, the unit cell of CeO2 increases. This is because the eight-coordinated Ce4+ cation size is about 1 Å, while the eight-coordinated Ce3+ cation size is about 1.1 Å. Substituting some of the Ce4+ cations with metal cations with the same formal oxidation state (M4+) but a smaller size compensates partly for the lattice expansion. This is particularly successful when Zr4+ cations are used (size ca. 0.8 Å) [7,8]. While the substitution is valid up to about 50% (maintaining the fluorite structure of CeO2) [9], phase segregation occurs at high temperatures (at 1000 °C or so) [10].
- Charge transfer: substitution with oxidizable higher-valence cations. In this case, a fraction of Ce4+ cations is substituted with metal cations that can donate electrons and themselves be oxidized [11]. The substitution of Ce4+ with U4+ was found to enhance the reduction of CeO2, particularly at low levels [12,13]. Upon the removal of an oxygen atom, three Ce3+ cations are formed (instead of two), and one U4+ cation is oxidized to a U5+ action. In addition, the fact that both oxides, CeO2 and UO2, have a fluorite structure and both cations have the same size makes them miscible for the entire ratio range [14]. The optimal dosing for the reduction of Ce cations is not clear yet, and neither is the temperature at which phase segregation occurs.
- Charge compensation (alio-valencies): lattice distortion. While the substitution of Ce4+ with metal cations of a lower oxidation state will create vacancies, these vacancies are not charged. In other words, there is no increase in electron charge. The effect is, however, clear; for example, the substitution of Ce4+ cations with Fe3+ cations (up to about 20%) results in a considerable reduction of the host oxide [15]. This is thought to be due to the distortion of the lattice structure, making it less stable and therefore enabling further reduction [16]. In recent work, this was found to be the case for high-temperature reduction (with no chemical input). Yet, considerable phase segregation occurred after one TCWS reaction cycle [17].
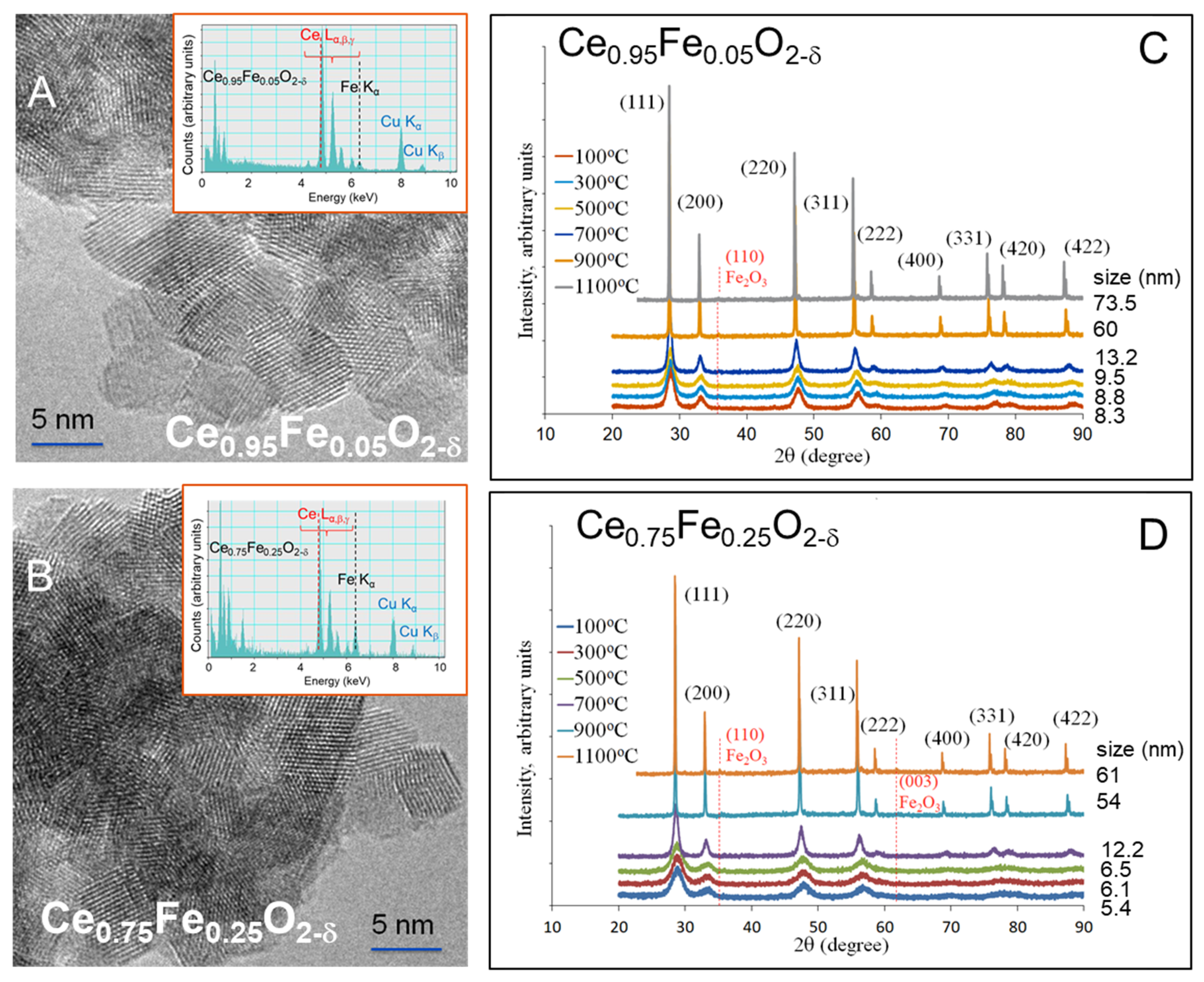
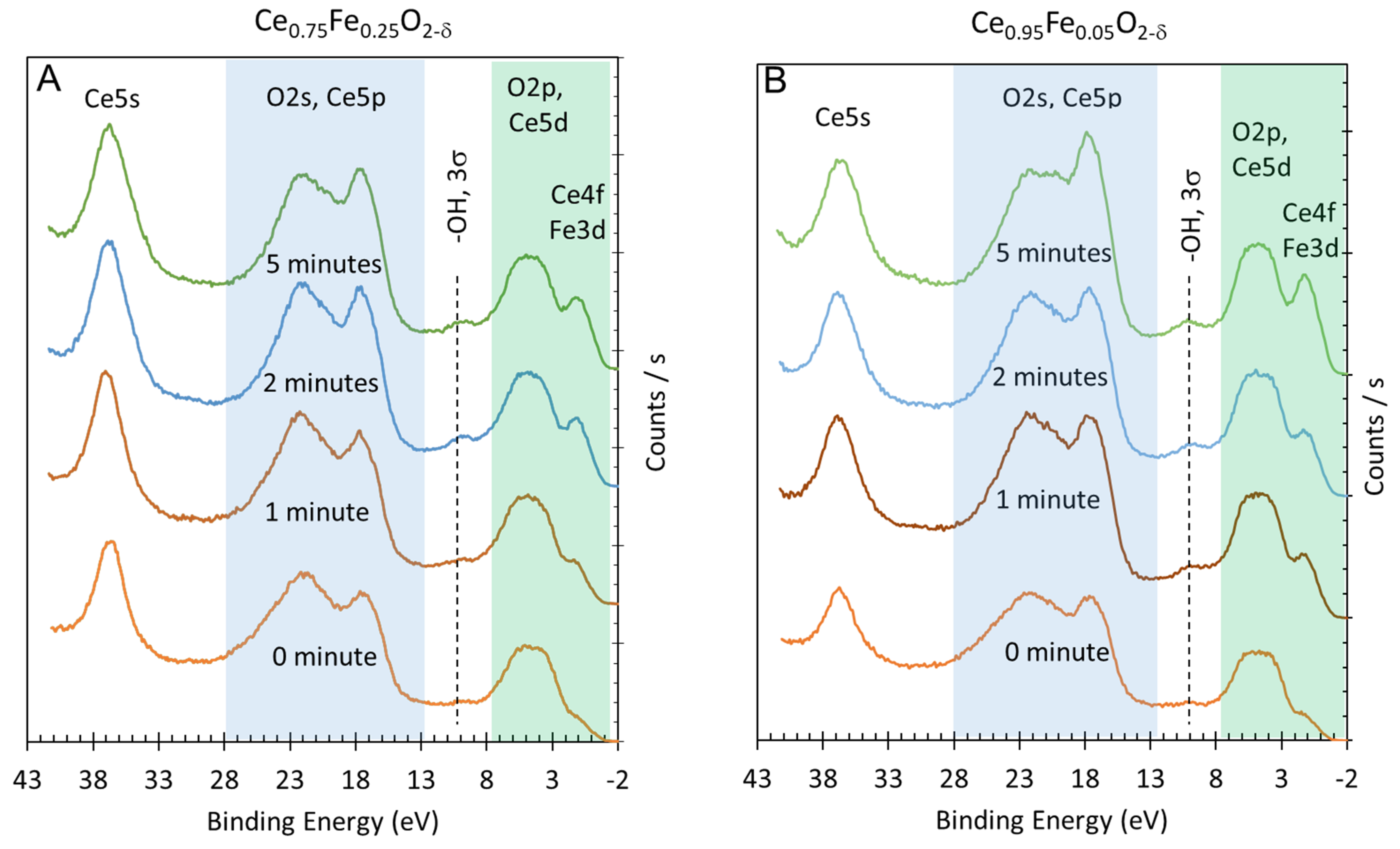
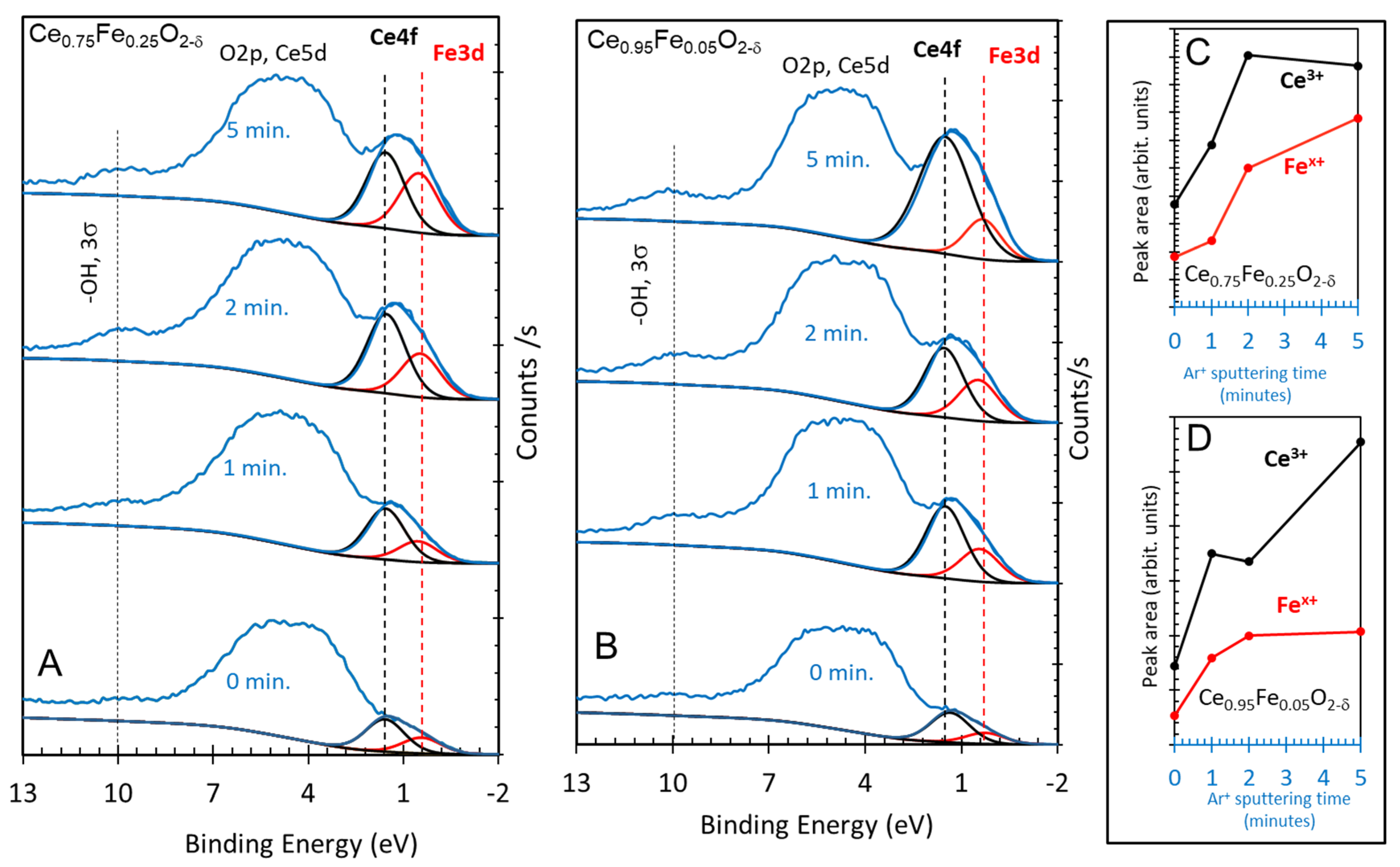
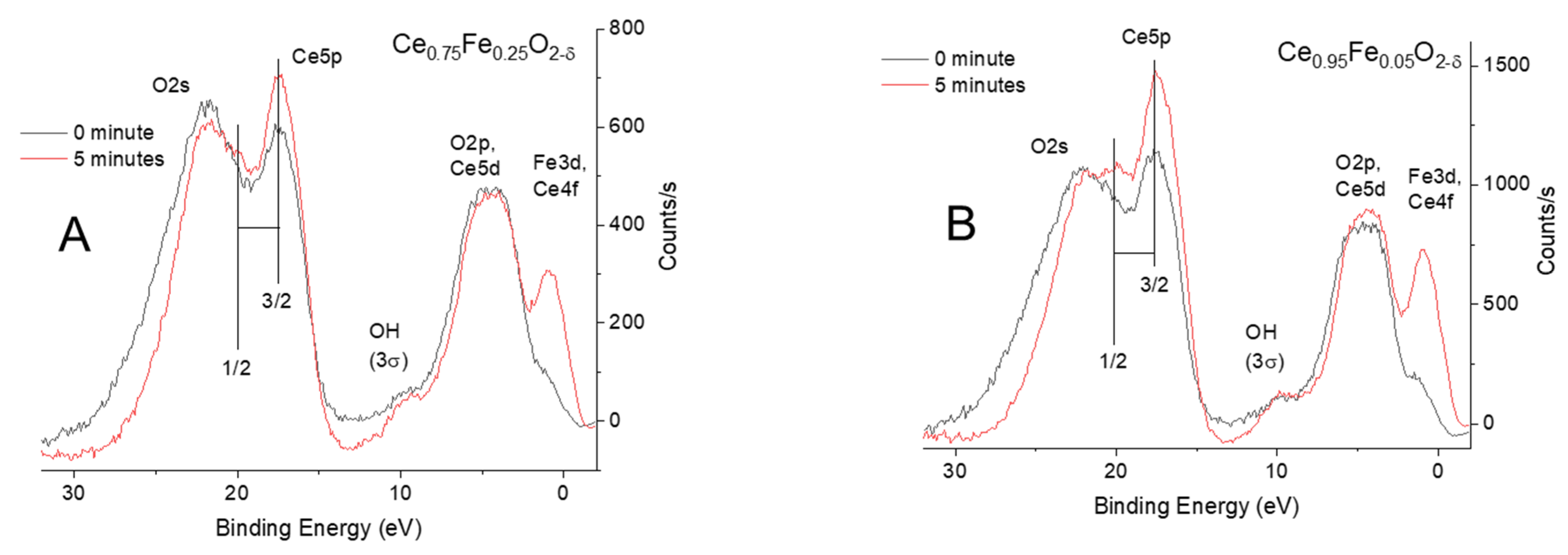
2. Results and Discussion
3. Experimental Section
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trovarelli, A. Catalytic Properties of Ceria and CeO2-Containing Materials. Catal. Rev. Sci. Eng. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Yang, C.; Lu, Y.; Zhang, L.; Kong, Z.; Yang, T.; Tao, L.; Zou, Y.; Wang, S. Defect Engineering on CeO2-Based Catalysts for Heterogeneous Catalytic Applications. Small Struct. 2021, 2, 2100058. [Google Scholar] [CrossRef]
- Torrente-Murciano, L.; Garcia-Garcia, R.R. Effect of nanostructured support on the WGSR activity of Pt/CeO2 catalysts. Catal. Commun. 2015, 71, 1–6. [Google Scholar] [CrossRef]
- Idriss, H. Oxygen vacancies role in thermally driven and photon driven catalytic reactions. Chem. Catal. 2022, 2, 1549–1560. [Google Scholar] [CrossRef]
- Onigbajumo, A.; Swarnkar, P.; Will, G.; Sundararajan, T.; Taghipour, A.; Couperthwaite, S.; Steinberg, T.; Rainey, T. Techno-economic evaluation of solar-driven ceria thermochemical water-splitting for hydrogen production in a fluidized bed reactor. J. Clean. Prod. 2022, 371, 133303. [Google Scholar] [CrossRef]
- Chueh, W.C.; Falter, C.; Abbott, M.; Scipio, D.; Furler, P.; Haile, S.M.; Steinfeld, A. High-flux solar-driven thermochemical dissociation of CO2 and H2O using nonstoichiometric ceria. Science 2010, 330, 797–801. [Google Scholar] [CrossRef]
- Alifanti, M.; Baps, B.; Blangenois, N.; Naud, J.; Grange, P.; Delmon, B. Characterization of CeO2-ZrO2 Mixed Oxides. Comparison of the Citrate and Sol-Gel Preparation Methods. Chem. Mater. 2003, 15, 395–403. [Google Scholar] [CrossRef]
- Andersson, D.A.; Simak, S.I.; Skorodumova, N.V.; Abrikosov, I.A.; Johansson, B. Redox properties of CeO2–MO2 (M = Ti, Zr, Hf, or Th) solid solutions from first principles calculations. Appl. Phys. Lett. 2007, 90, 031909. [Google Scholar] [CrossRef]
- Diagne, C.; Idriss, H.; Kiennemann, A. Hydrogen Production by Ethanol Reforming over Rh/CeO2-ZrO2 Catalysts. Catal. Commun. 2002, 3, 565–571. [Google Scholar] [CrossRef]
- Grau-Crespo, R.; de Leeuw, N.H.; Hamad, S.; Waghmare, U.V. Phase separation and surface segregation in ceria–zirconia solid solutions. Proc. R. Soc. A 2011, 467, 1925–1938. [Google Scholar] [CrossRef]
- Hanken, B.E.; Stanek, C.R.; Grønbech-Jensen, N.; Asta, M. Computational study of the energetics of charge and cation mixing in U1−xCexO2. Phys. Rev. B 2011, 84, 085131. [Google Scholar] [CrossRef]
- Scaranto, G.; Idriss, H. The Effect of Uranium Cations on the Redox Properties of CeO2 Within the Context of Hydrogen Production from Water. Top. Catal. 2015, 58, 143–148. [Google Scholar] [CrossRef]
- Al-Shankiti, I.; Al-Otaibi, F.; Al-Salik, Y.; Idriss, H. Solar thermal hydrogen production from water over modified CeO2 materials. Top. Catal. 2013, 56, 1129–1138. [Google Scholar] [CrossRef]
- Prieur, D.; Vigier, J.-F.; Popa, K.; Walter, O.; Dieste, O.; Varga, Z.; Beck, A.; Vitova, T.; Scheinost, A.; Martin, P. Charge distribution in U1−xCexO2+y nanoparticles. Inorg. Chem. 2021, 60, 14550–14556. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Waghmare, U.V.; Hegde, M.S. Correlation of Oxygen Storage Capacity and Structural Distortion in Transition-Metal-, Noble-Metal-, and Rare-Earth-Ion-Substituted CeO2 from First Principles Calculation. Chem. Mater. 2010, 22, 5184–5198. [Google Scholar] [CrossRef]
- Gupta, A.; Kumar, A.; Waghmare, U.V.; Hegde, M.S. Origin of activation of Lattice Oxygen and Synergistic Interaction in Bimetal-Ionic Ce0.89Fe0.1Pd0.01O2-δ Catalyst. Chem. Mater. 2009, 21, 4880–4891. [Google Scholar] [CrossRef]
- Al-Taweel, S.; Nadeem, M.A.; Idriss, H. A study of CexFe1−xO2 as a reducible oxide for the thermal hydrogen production from water. Energy Technol. 2022, 10, 2100491. [Google Scholar] [CrossRef]
- Cheng, L.; Yue, X.; Fan, J.; Xiang, Q. Site-Specific Electron-Driving Observations of CO2-to-CH4 Photoreduction on Co-Doped CeO2/Crystalline Carbon Nitride S-Scheme Heterojunctions. Adv. Mater. 2022, 34, 2200929. [Google Scholar] [CrossRef]
- Zhao, S.; Kang, D.; Liu, Y.; Wen, Y.; Xie, X.; Yi, H.; Tang, X. Spontaneous Formation of Asymmetric Oxygen Vacancies in Transition-Metal-Doped CeO2 Nanorods with Improved Activity for Carbonyl Sulfide Hydrolysis. ACS Catal. 2020, 10, 11739–11750. [Google Scholar] [CrossRef]
- He, W.; Ran, J.; Yang, G.; He, Z.; Huang, X.; Crittenden, J. Promoting effect of Co-doped CeO2 nanorods activity and SO2 resistance for Hg0 removal. Fuel 2022, 317, 123320. [Google Scholar] [CrossRef]
- Ahn, K.; Yoo, D.S.; Prasad, D.H.; Lee, H.-W.; Chung, Y.-C.; Lee, J.-H. Role of Multivalent Pr in the Formation and Migration of Oxygen Vacancy in Pr-Doped Ceria: Experimental and First-Principles Investigations. Chem. Mater. 2012, 24, 4261–4267. [Google Scholar] [CrossRef]
- Zhang, M.; Jiang, D.; Jiang, H. Enhanced oxygen storage capacity of Ce0.88Mn0.12Oy compared to CeO2: An experimental and theoretical investigation. Mater. Res. Bull. 2012, 47, 4006–4012. [Google Scholar] [CrossRef]
- Krcha, M.D.; Janik, M.J. Examination of Oxygen Vacancy Formation in Mn-Doped CeO2 (111) Using DFT+U and the Hybrid Functional HSE06. Langmuir 2013, 29, 10120–10131. [Google Scholar] [CrossRef] [PubMed]
- Righi, G.; Benedetti, S.; Rita Magri, R. Investigation of the structural and electronic differences between silver and copper doped ceria using the density functional theory. J. Phys. Condens. Matter. 2022, 34, 204010. [Google Scholar] [CrossRef] [PubMed]
- Nolan, M. Enhanced oxygen vacancy formation in ceria (111) and (110) surfaces doped with divalent cations. J. Mater. Chem. 2011, 21, 9160–9168. [Google Scholar] [CrossRef]
- Wang, X.; Shen, M.; Wang, J.; Fabris, S. Enhanced Oxygen Buffering by Substitutional and Interstitial Ni Point Defects in Ceria: A First-Principles DFT+U Study. J. Phys. Chem. C 2010, 114, 10221–10228. [Google Scholar] [CrossRef]
- Grieshammer, S.; Grope, B.O.H.; Koettgen, J.; Martin, M. A combined DFT + U and Monte Carlo study on rare earth doped ceria. Phys. Chem. Chem. Phys. 2014, 16, 9974–9986. [Google Scholar] [CrossRef]
- Wang, H.; Qu, Z.; Xie, H.; Maeda, N.; Miao, L.; Wang, Z. Insight into the mesoporous FexCe1-xO2-x catalysts for selective catalytic reduction of NO with NH3: Regulable structure and activity. J. Catal. 2016, 338, 56–67. [Google Scholar] [CrossRef]
- Liu, X.-S.; Wang, X.-D.; Yao, M.; Cui, W.; Yan, H. Effects of Fe doping on oxygen vacancy formation and CO adsorption and oxidation at the ceria(111) surface. Catal. Commun. 2015, 63, 35–40. [Google Scholar] [CrossRef]
- Lykaki, M.; Stefa, S.; Carabineiro, S.A.C.; Pandis, P.K.; Stathopoulos, V.N.; Konsolakis, M. Facet-Dependent Reactivity of Fe2O3/CeO2 Nanocomposites: Effect of Ceria Morphology on CO Oxidation. Catalysts 2019, 9, 371. [Google Scholar] [CrossRef]
- Kaneko, H.; Miura, T.; Ishihara, H.; Taku, S.; Yokoyama, T.; Nakajima, H.; Tamaura, Y. Reactive ceramics of CeO2−xMOx (M = Mn, Fe, Ni, Cu) for H2 generation by two-step water splitting using concentrated solar thermal energy. Energy 2007, 32, 656–663. [Google Scholar] [CrossRef]
- Liang, C.; Ma, Z.; Lin, H.; Ding, L.; Qiu, J.; Frandsen, W.; Su, D. Template preparation of nanoscale CexFe1−xO2 solid solutions and their catalytic properties for ethanol steam reforming. J. Mater. Chem. 2009, 19, 1417–1424. [Google Scholar] [CrossRef]
- Metal Oxide Nanostructures Chemistry: Synthesis from Aqueous Solutions, 2nd ed.; Jolivet, J.-P. (Ed.) Chapter 5, Surface Chemistry and Physicochemistry of Oxides; Oxford University Press (2015) Editions EDP Sciences: Oxford, UK, 2015; pp. 181–226. [Google Scholar] [CrossRef]
- Maslakov, K.I.; Teterin, Y.A.; Popel, A.J.; Teterin, A.Y.; Ivanov, K.E.; Kalmykov, S.N.; Petrov, V.G.; Petrov, P.K.; Farnan, I. XPS study of ion irradiated and unirradiated CeO2 bulk and thin film Samples. Appl. Surf. Sci. 2018, 448, 154–162. [Google Scholar] [CrossRef]
- Maslakov, K.I.; Teterin, Y.A.; Ryzhkov, M.V.; Ivanov, K.E.; Popel, A.J.; Teterin, A.Y.; Kalmykov, S.N.; Petrov, V.G.; Petrov, P.K.; Farnan, I. The electronic structure and the nature of the chemical bond in CeO2. Phys. Chem. Chem. Phys. 2018, 20, 16167–16175. [Google Scholar] [CrossRef]
- Eck, S.; Castellarin-Cudia, C.; Surnev, S.; Ramsey, M.G.; Netzer, F.P. Growth and thermal properties of ultrathin cerium oxide layers on Rh(111). Surf. Sci. 2002, 520, 173–185. [Google Scholar] [CrossRef]
- Soria, E.; Román, E.; Williams, E.M.; de Segovia, J.L. Observations with synchrotron radiation (20–120 eV) of the TiO2(110)–glycine interface. Surf. Sci. 1999, 433–435, 543–548. [Google Scholar] [CrossRef]
- Parkinson, G.S. Iron oxide surfaces. Surf. Sci. Rep. 2016, 71, 272–365. [Google Scholar] [CrossRef]
- Mansour, A.N.; Brizzolara, R.A. Characterization of the Surface of FeO Powder by XPS. Surf. Sci. Spectra 1996, 4, 345–350. [Google Scholar] [CrossRef]
- Preisinger, M.; Krispin, M.; Rudolf, T.; Horn, S.; Strongin, D.R. Electronic structure of nanoscale iron oxide particles measured by scanning tunneling and photoelectron spectroscopies. Phys. Rev. B 2005, 71, 165409. [Google Scholar] [CrossRef]
- Radu, T.; Iacovita, C.; Benea, D.; Turcu, R. X-Ray Photoelectron Spectroscopic Characterization of Iron Oxide Nanoparticles. Appl. Surf. Sci. 2017, 405, 337–343. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Kobe, B.A.; McIntyre, N.S. Studies of the oxidation of iron by water vapour using X-ray photoelectron spectroscopy and QUASES. Surf. Sci. 2004, 572, 217–227. [Google Scholar] [CrossRef]
- Temesghen, W.; Sherwood, P.M.A. Analytical utility of valence band X-ray photoelectron spectroscopy of iron and its oxides, with spectral interpretation by cluster and band structure calculations. Anal. Bioanal. Chem. 2002, 373, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Bagus, P.S.; Nelin, C.J.; Brundle, C.R.; Vincent Crist, C.B.; Lahiri, N.; Rosso, K.M. Combined multiplet theory and experiment for the Fe 2p and 3p XPS of FeO and Fe2O3. J. Chem. Phys. 2021, 154, 094709. [Google Scholar] [CrossRef] [PubMed]
- Fondell, M.; Gorgoi, M.; Boman, M.; Lindblad, A. Surface modification of iron oxides by ion bombardment—Comparing depth profiling by HAXPES and Ar ion sputtering. J. Electron Spectrosc. Relat. Phenom. 2018, 224, 23–26. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
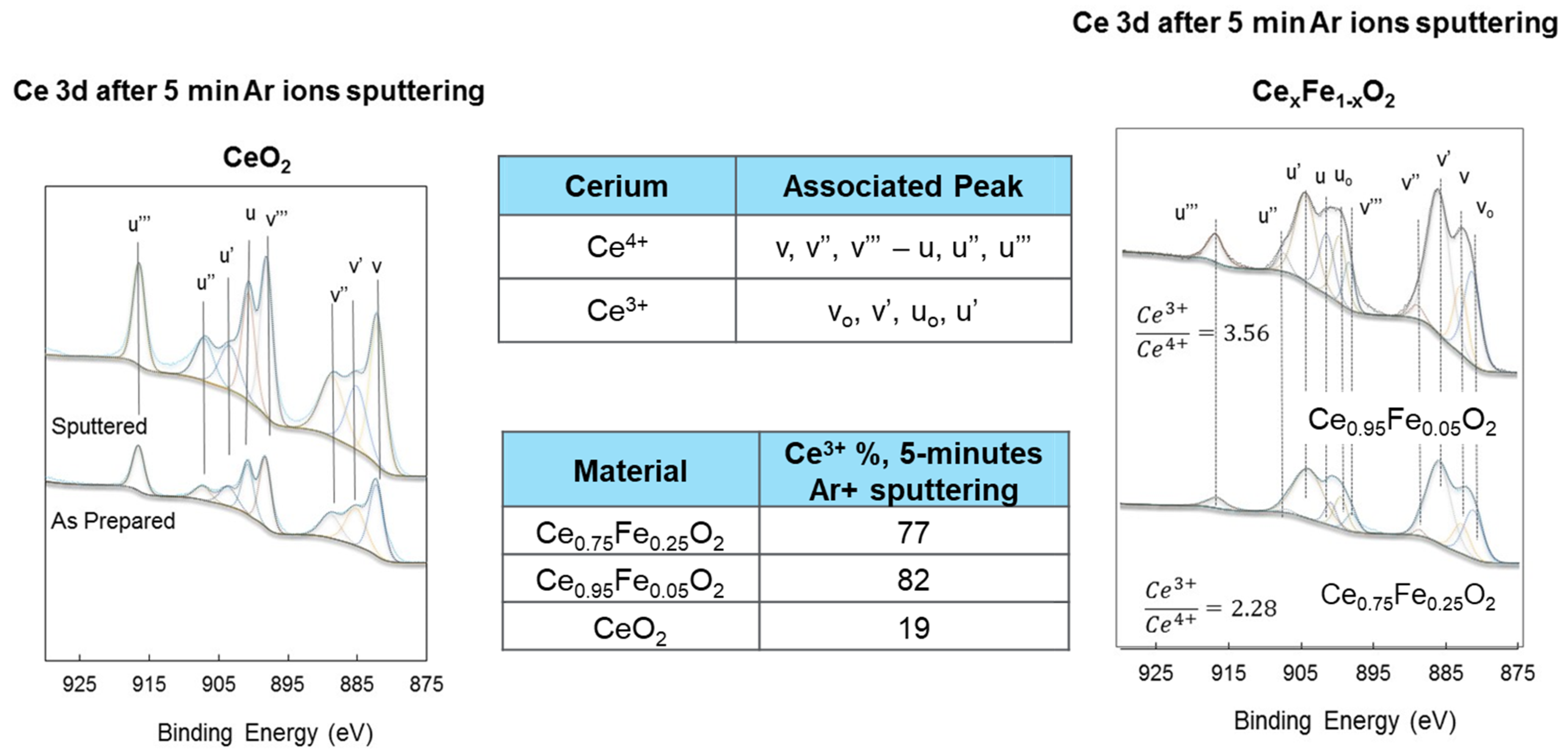
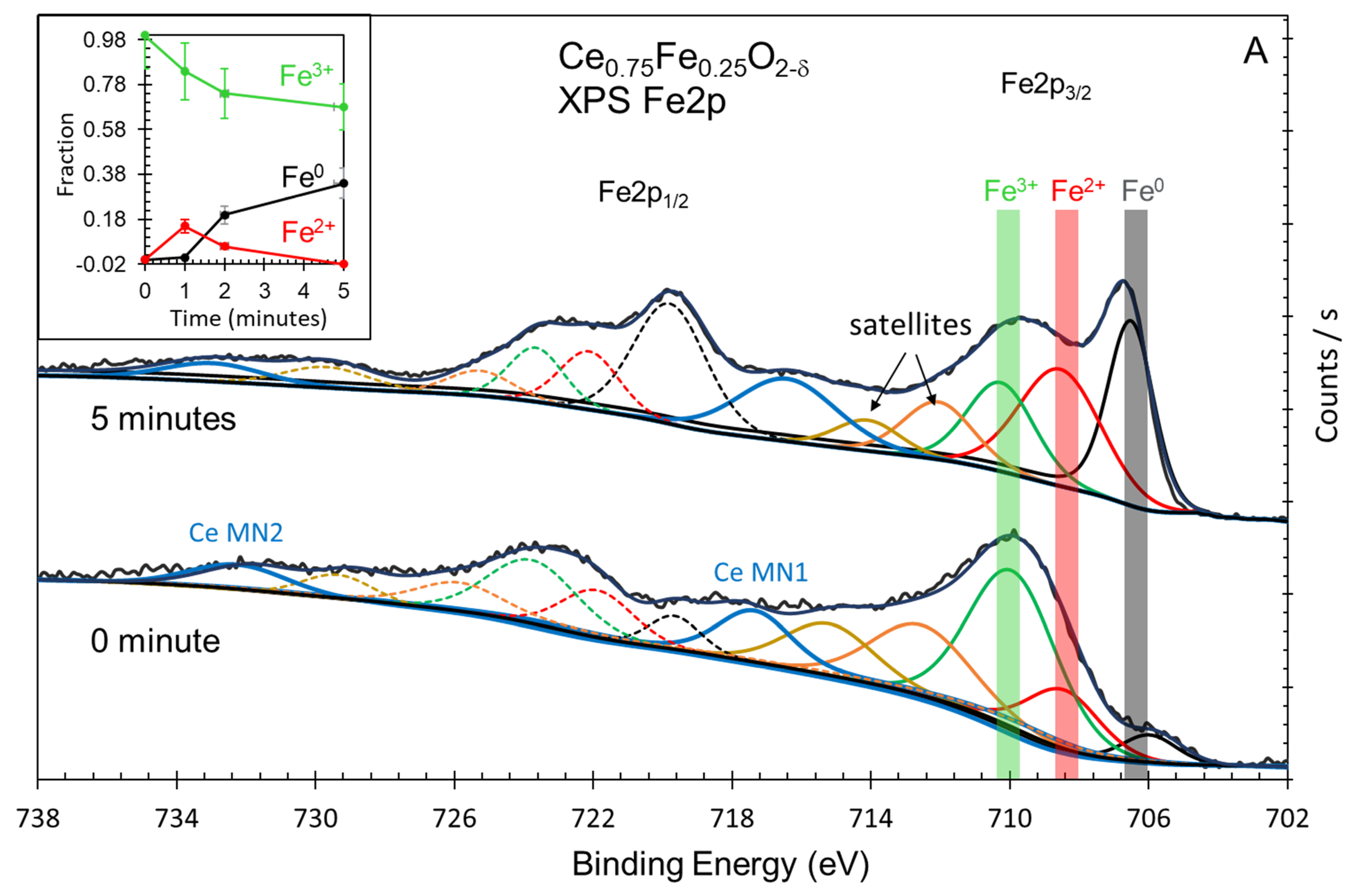
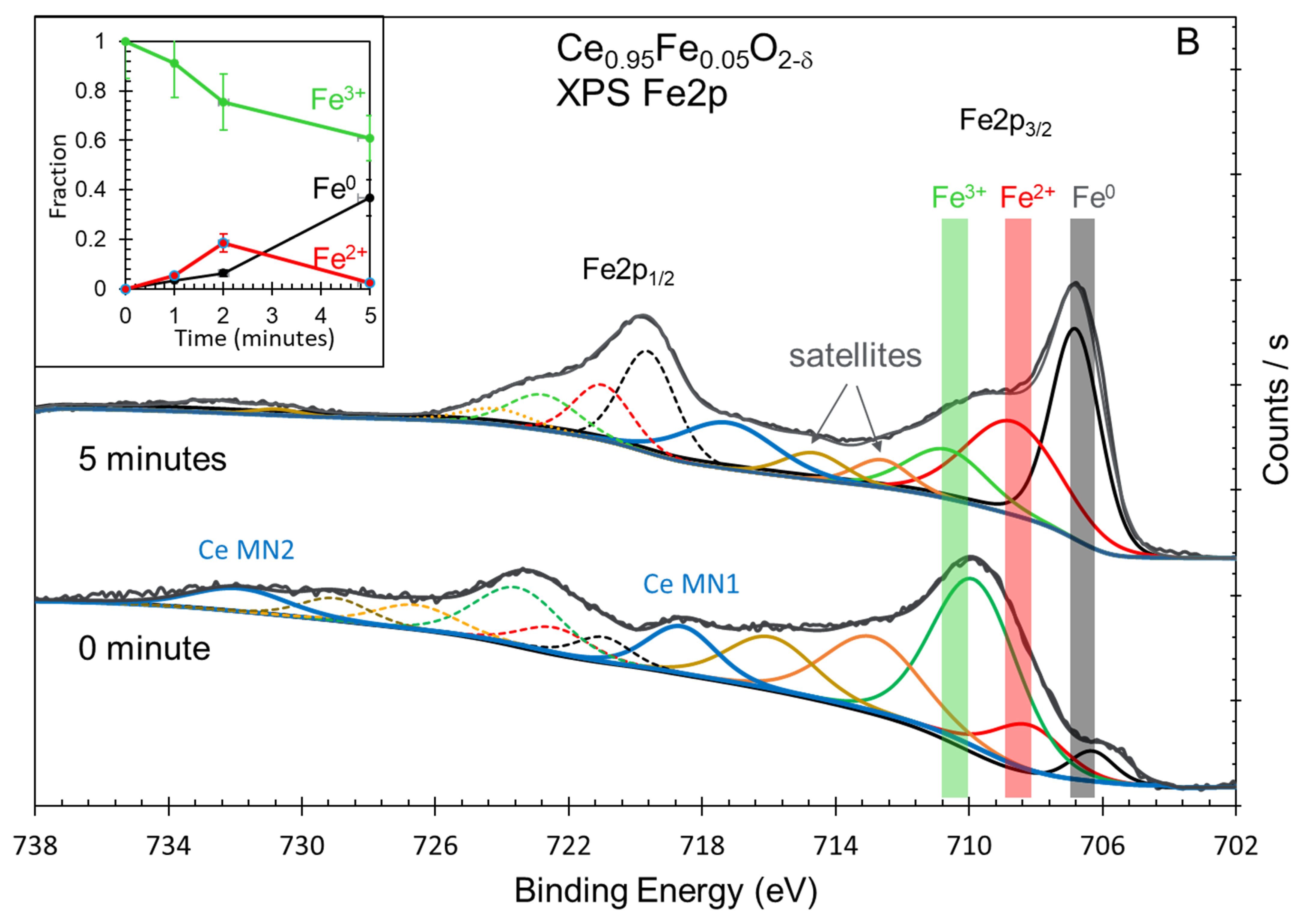
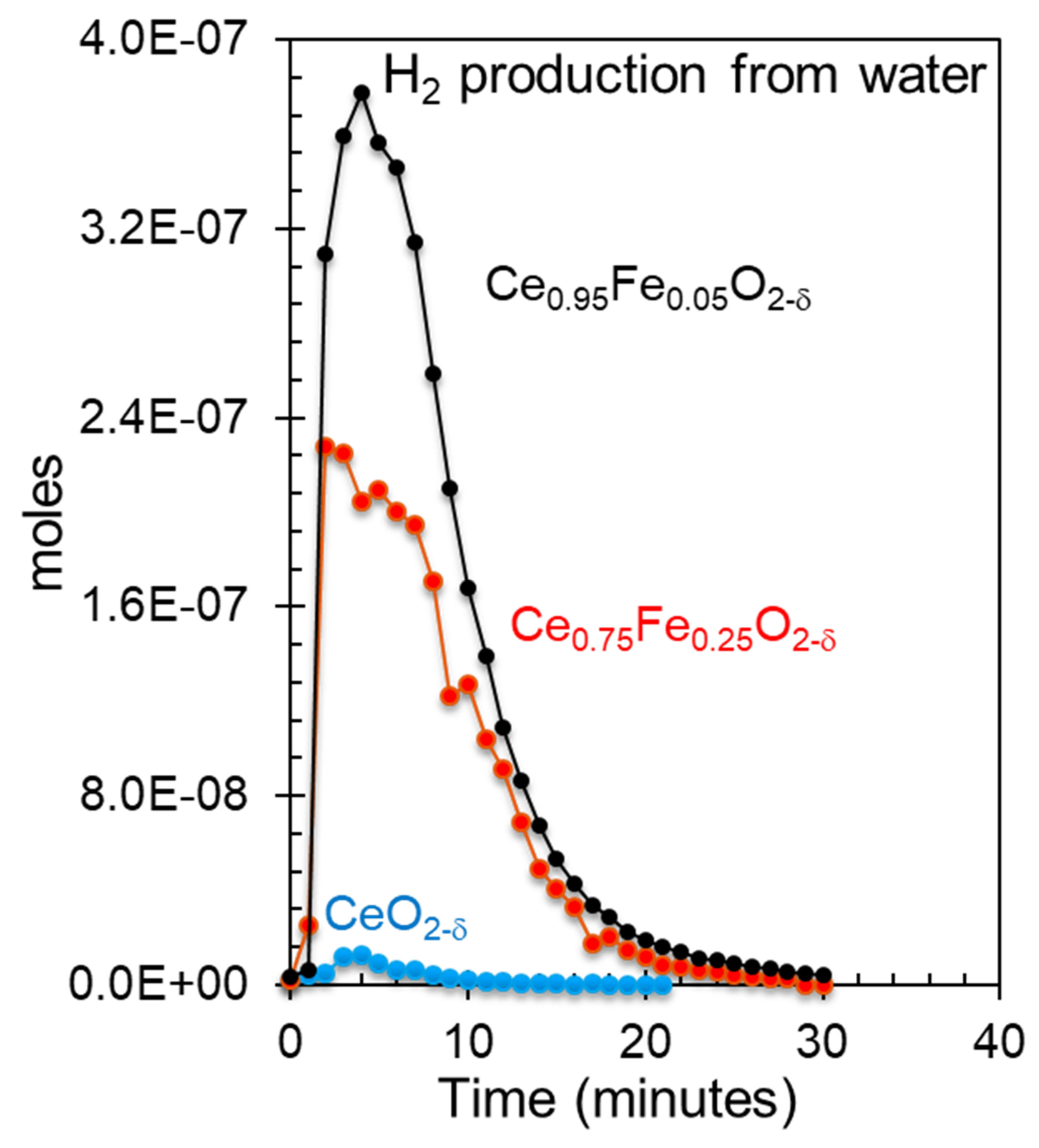
| Oxide | Ce3+/Ce4+ (Ce3d) | [Ce4f + Fe3dx]/O2p | Fe0/Fe3+ (Fe2p) | Ce5p/O2s | H2 Production (mol/g) |
|---|---|---|---|---|---|
| CeO2 | 0.2 | - | - | 0 | 0.2 × 10−6 |
| Ce0.75Fe0.25O2-δ | 2.3 | 0.3 | 0.5 | 1.6 | 7.4 × 10−6 |
| Ce0.95Fe0.05O2-δ | 3.6 | 0.4 | 0.6 | 1.75 | 11.4 × 10−6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idriss, H. A Core and Valence-Level Spectroscopy Study of the Enhanced Reduction of CeO2 by Iron Substitution—Implications for the Thermal Water-Splitting Reaction. Inorganics 2024, 12, 42. https://doi.org/10.3390/inorganics12020042
Idriss H. A Core and Valence-Level Spectroscopy Study of the Enhanced Reduction of CeO2 by Iron Substitution—Implications for the Thermal Water-Splitting Reaction. Inorganics. 2024; 12(2):42. https://doi.org/10.3390/inorganics12020042
Chicago/Turabian StyleIdriss, Hicham. 2024. "A Core and Valence-Level Spectroscopy Study of the Enhanced Reduction of CeO2 by Iron Substitution—Implications for the Thermal Water-Splitting Reaction" Inorganics 12, no. 2: 42. https://doi.org/10.3390/inorganics12020042
APA StyleIdriss, H. (2024). A Core and Valence-Level Spectroscopy Study of the Enhanced Reduction of CeO2 by Iron Substitution—Implications for the Thermal Water-Splitting Reaction. Inorganics, 12(2), 42. https://doi.org/10.3390/inorganics12020042