The Silacyclobutene Ring: An Indicator of Triplet State Baird-Aromaticity


Abstract
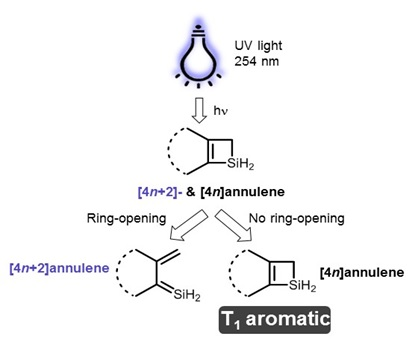
: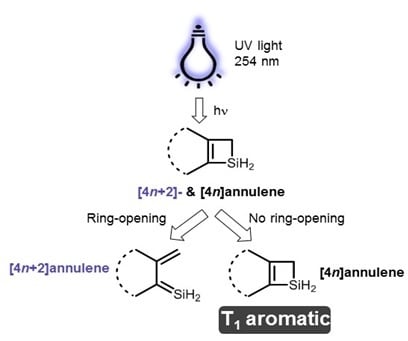
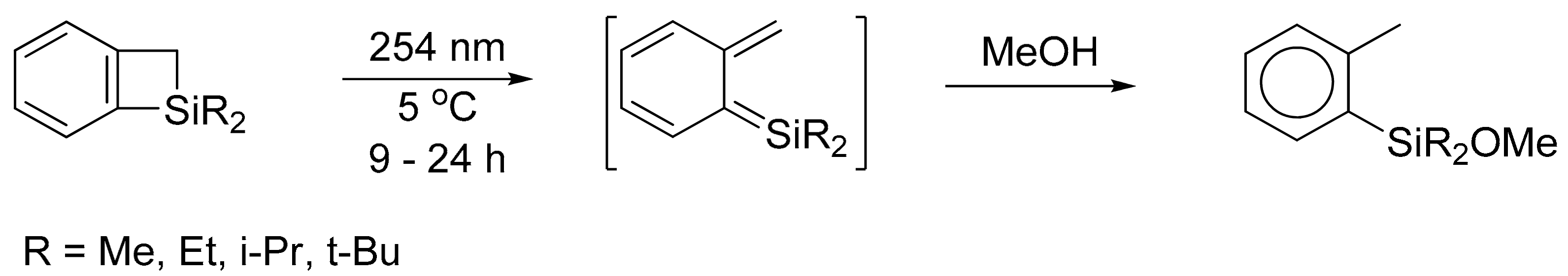
1. Introduction
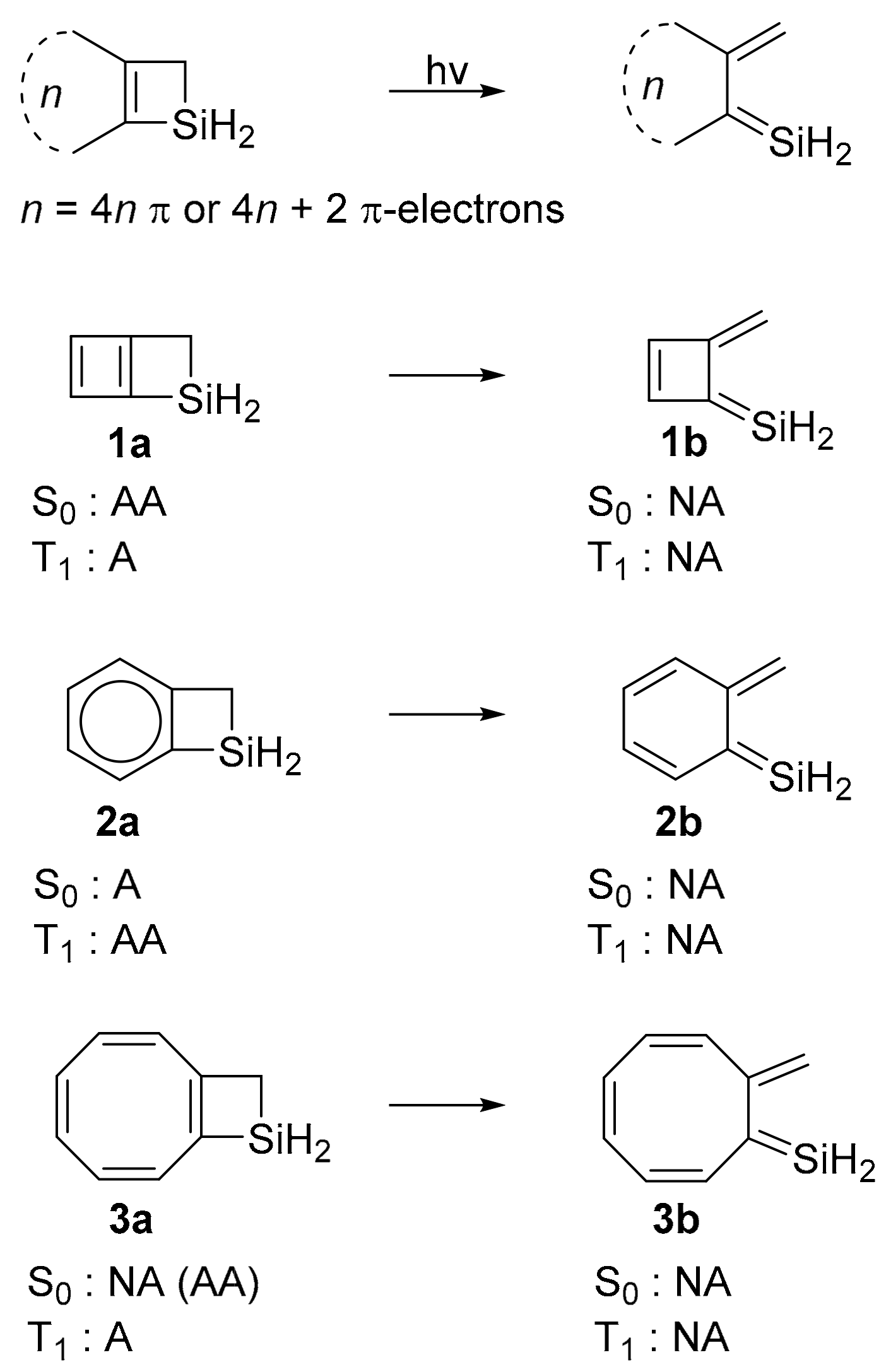
2. Results and Discussion
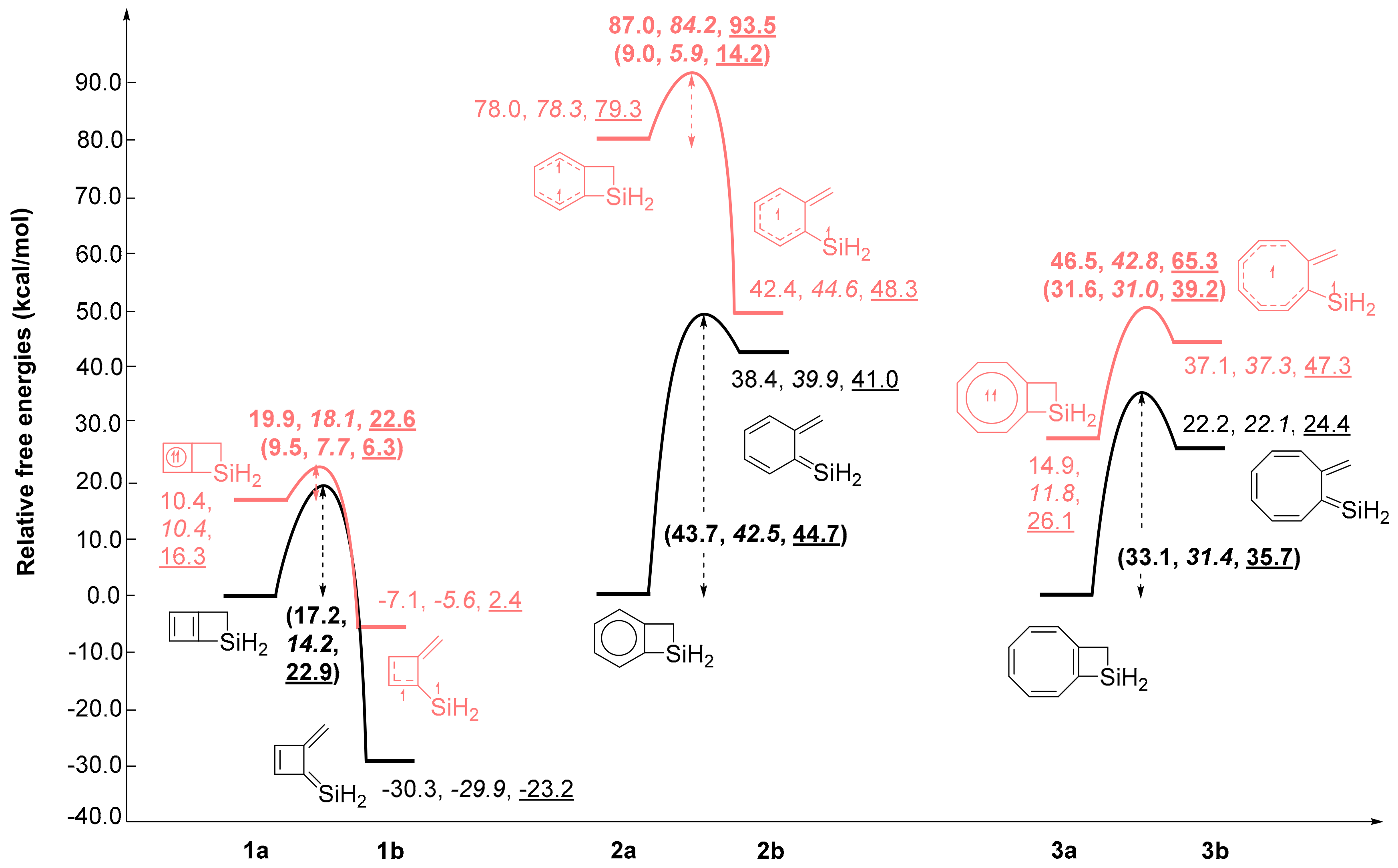
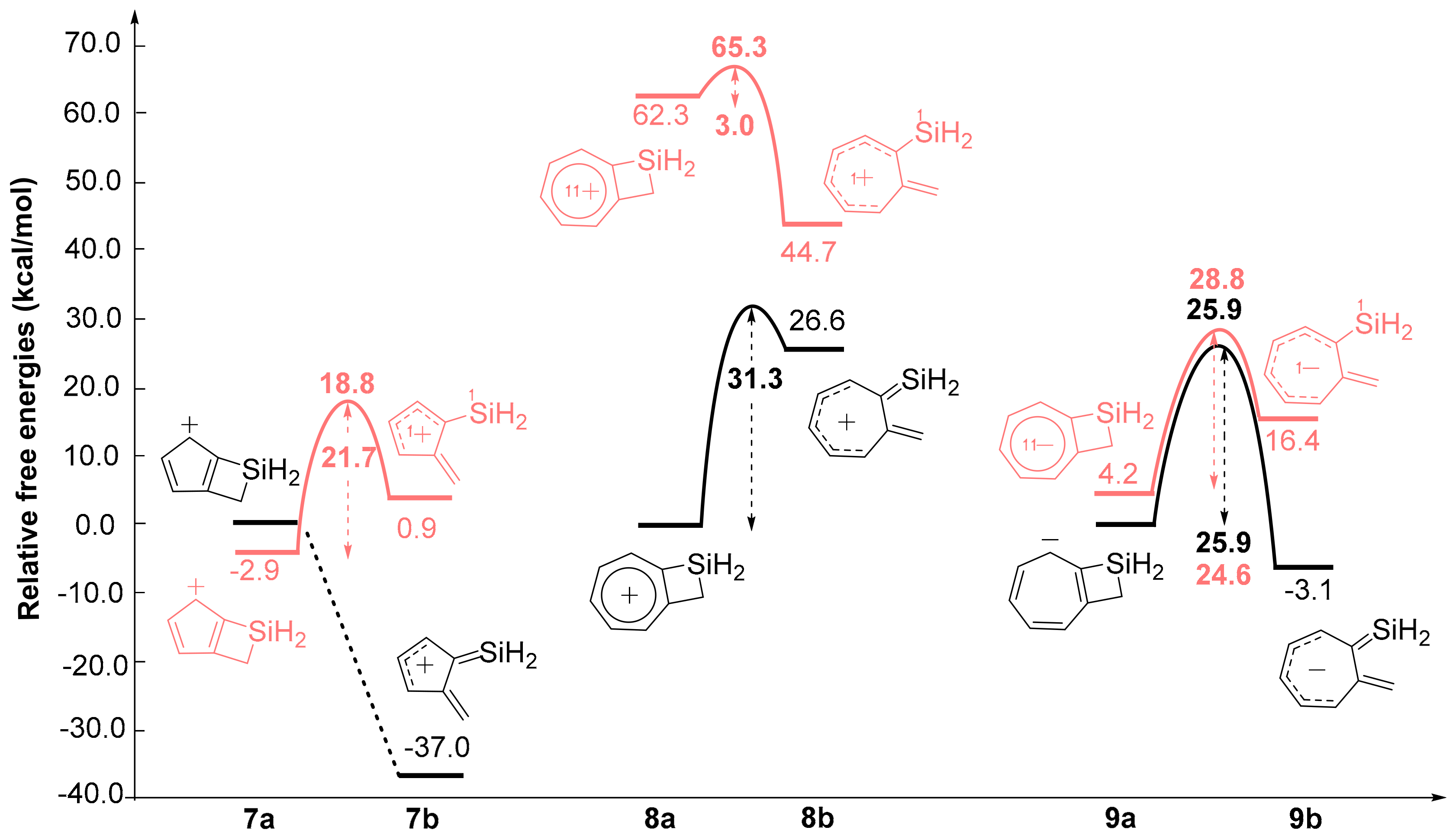
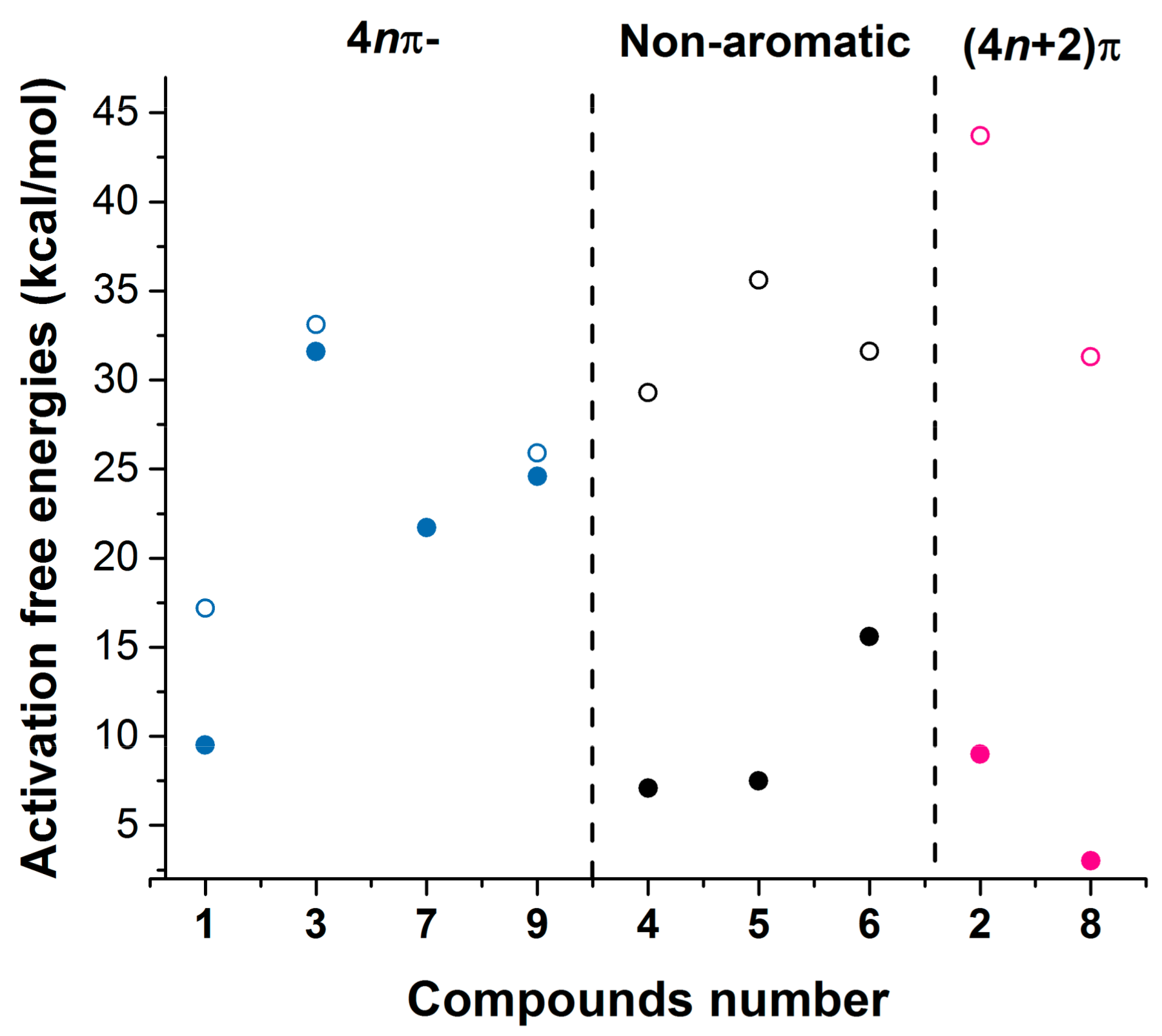
2.1. Energy Changes
2.2. Changes in T1 State (Anti)aromaticity upon Ring-Opening
2.2.1. Harmonic Oscillator Model of Aromaticity (HOMA) Values
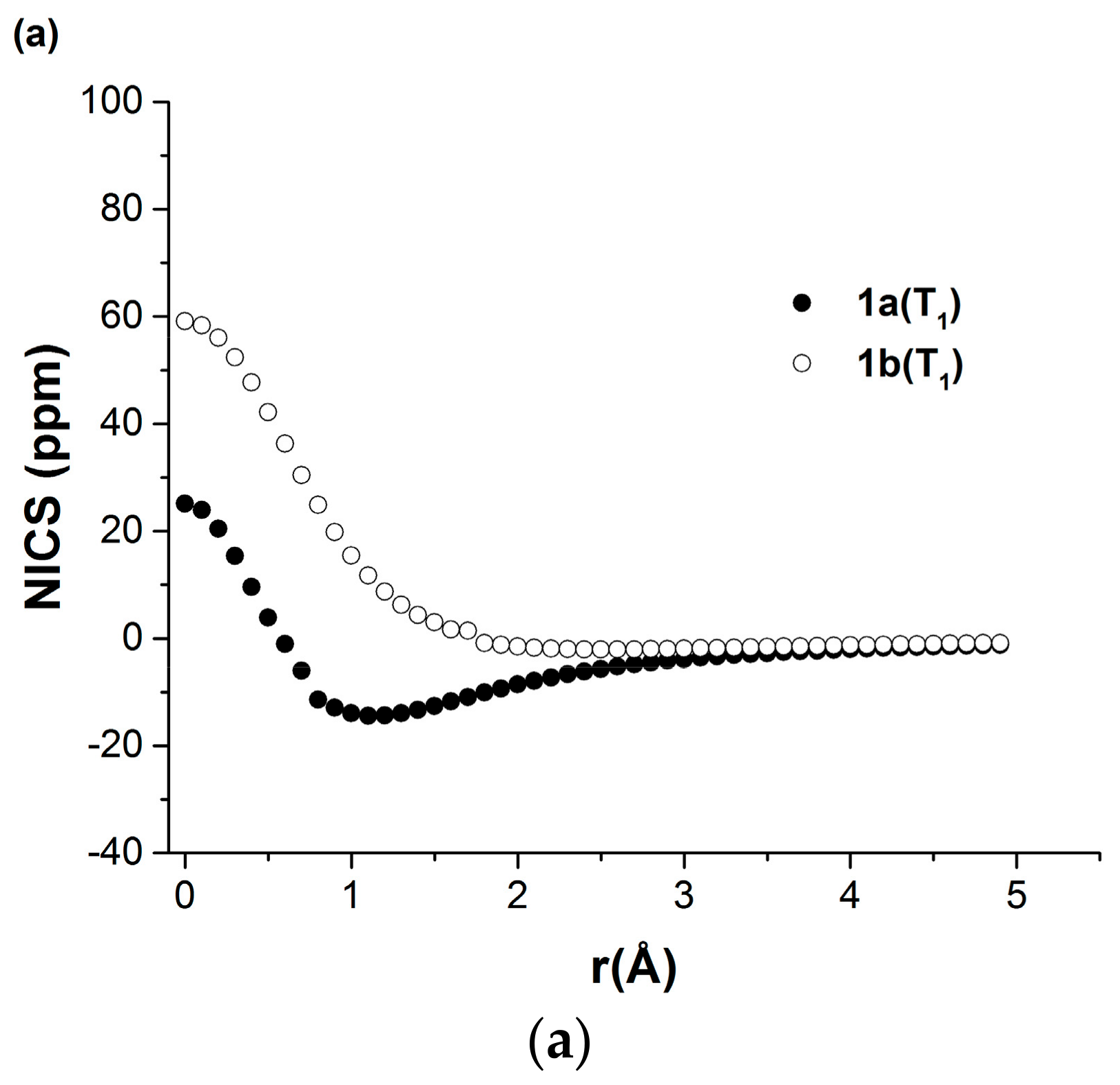
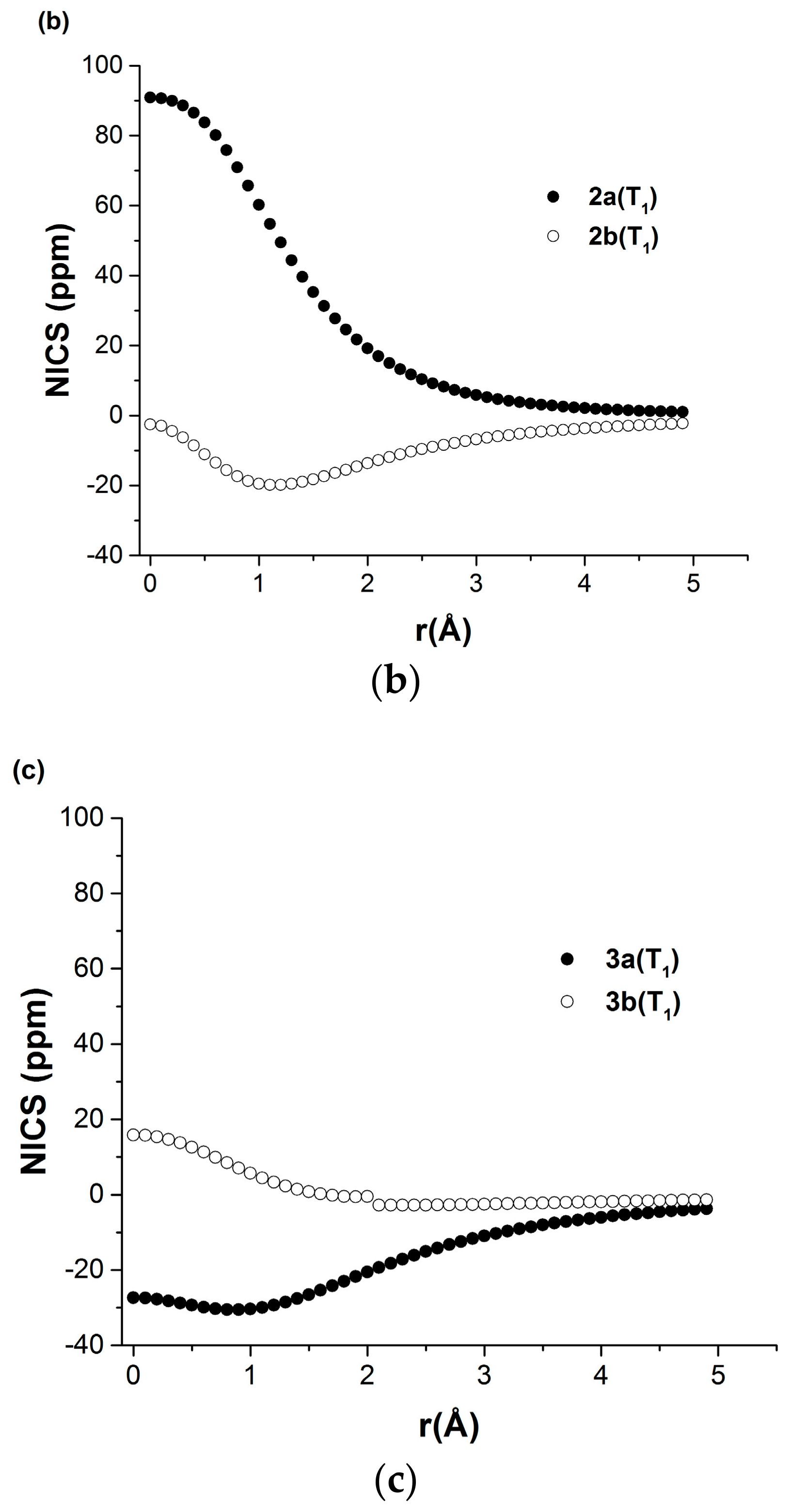
2.2.2. Nucleus Independent Chemical Shift (NICS) Scans
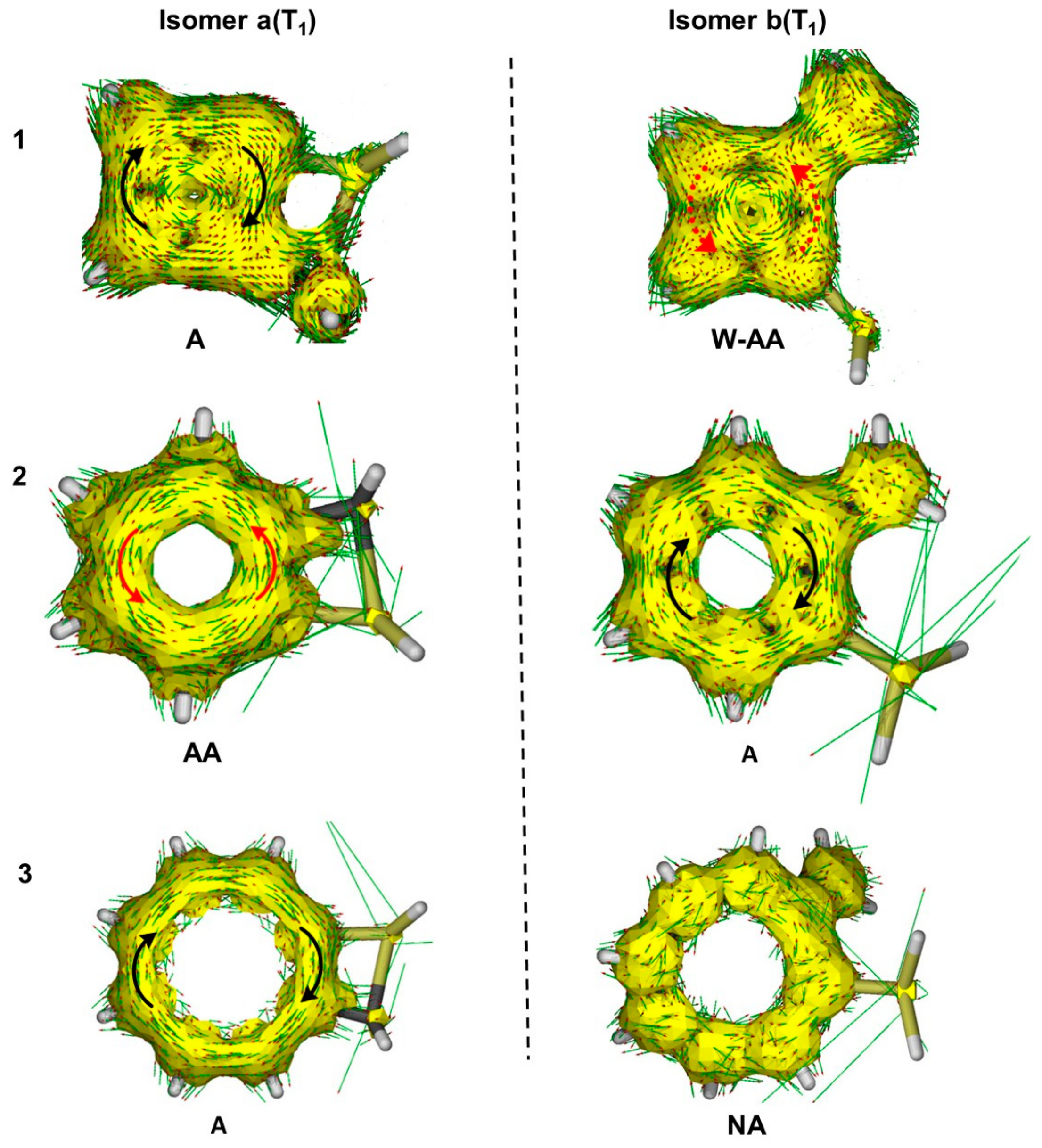
2.2.3. Anisotropy of the Induced Current Density (ACID) Plots
2.2.4. Isomerization Stabilization Energy (ISE) Values
2.3. Five- and Seven-Membered Carbocyclic Anions and Cations Fused to SCB
2.4. Polycyclic Structural Units Fused to SCB
3. Computational Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baird, N.C. Quantum Organic Photochemistry. II. Resonance and Aromaticity in the Lowest 3ππ* State of Cyclic Hydrocarbons. J. Am. Chem. Soc. 1972, 94, 4941–4948. [Google Scholar] [CrossRef]
- Ottosson, H. Organic Photochemistry: Exciting Excited State Aromaticity. Nat. Chem. 2012, 4, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.; Dahlstrand, C.; Kilså, K.; Ottosson, H. Excited State Aromaticity and Antiaromaticity: Oppertunities for Photophysical and Photochemical Rationalizations. Chem. Rev. 2014, 114, 5379–5425. [Google Scholar] [CrossRef] [PubMed]
- Gogonea, V.; Schleyer, P.R.; Schreiner, P.R. Consequences of Triplet Aromaticity in 4nπ-Electron Annulenes: Calculation of Magnetic Shieldings for Open-Shell Species. Angew. Chem. Int. Ed. 1998, 37, 1945–1948. [Google Scholar] [CrossRef]
- Glukhovtsev, M.N.; Reindl, B.; Schleyer, P.R. What is the Preferred Structure of the Singlet Cyclopentadienyl Cation? Mendeleev Commun. 1993, 3, 100–102. [Google Scholar] [CrossRef]
- Glukhovtsev, M.N.; Bach, R.D.; Laiter, S. Computational Study of the Thermochemistry of C5H5+ Isomers: Which C5H5+ Isomer is the Most Stable? J. Phys. Chem. 1996, 100, 10952–10955. [Google Scholar] [CrossRef]
- Mauksch, M.; Tsogoeva, S.B. A New Architecture for High Spin Organics based on Baird’s rule of 4n Electron Triplet Aromatics. Phys. Chem. Chem. Phys. 2017, 19, 4688–4694. [Google Scholar] [CrossRef] [PubMed]
- Karadakov, P.B. Ground- and Excited-State Aromaticity and Antiaromaticity in Benzene and Cyclobutadiene. J. Phys. Chem. A 2008, 112, 7303–7309. [Google Scholar] [CrossRef] [PubMed]
- Karadakov, P.B. Aromaticity and Antiaromaticity in the Low-Lying Electronic States of Cyclooctatetraene. J. Phys. Chem. A 2008, 112, 12707–12713. [Google Scholar] [CrossRef] [PubMed]
- Karadakov, P.B.; Hearnshaw, P.; Horner, K.E. Magnetic Shielding, Aromaticity, Antiaromaticity, and Bonding in the Lowest Lying Electronic States of Benzene and Cyclobutadiene. J. Org. Chem. 2016, 81, 11346–11352. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, M. Magnetic Susceptibility and Aromaticity in the Excited States of Benzene. J. Chem. Res. 2004, 2004, 573–574. [Google Scholar] [CrossRef]
- Haas, Y.; Zilberg, S. The ν14(b2u) Mode of Benzene in S0 and S1 and the Distortive Nature of the π Electron System: Theory and Experiment. J. Am. Chem. Soc. 1995, 117, 5387–5388. [Google Scholar] [CrossRef]
- Feixas, F.; Vandenbussche, J.; Bultinck, P.; Matito, E.; Solà, M. Electron Delocalization and Aromaticity in Low-Lying Excited States of Archetypal Organic Compounds. Phys. Chem. Chem. Phys. 2011, 13, 20690–20703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadakis, R.; Ottosson, H. The Excited State Antiaromatic Benzene Ring: A Molecular Mr Hyde? Chem. Soc. Rev. 2015, 44, 6472–6493. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.L. Strange Case of Dr. Jekyll and Mr. Hyde; Longmans, Green and Co.: London, UK, 1886; ISBN 978-0-553-21277-8. [Google Scholar]
- Aihara, J.-I. Aromaticity-Based Theory of Pericyclic Reactions. Bull. Chem. Soc. Jpn. 1978, 51, 1788–1792. [Google Scholar] [CrossRef]
- Fratev, F.; Monev, V.; Janoschek, R. Ab Initio Study of Cyclobutadiene in Excited States: Optimized Geometries, Electronic Tranisitions and Aromaticities. Tetrahedron 1982, 38, 2929–2932. [Google Scholar] [CrossRef]
- Garavelli, M.; Bernardi, F.; Cembran, A.; Castaño, O.; Frutos, L.S.; Merchán, M.; Olivucci, M. Cyclooctatetraene Computational Photo- and Thermal Chemistry: A Reactivity Model for Conjugated Hydrocarbons. J. Am. Chem. Soc. 2002, 124, 13770–13789. [Google Scholar] [CrossRef] [PubMed]
- Villaume, S.; Fogarty, H.A.; Ottosson, H. Triplet-State Aromaticity of 4nπ-Electron Monocycles: Analysis of Bifurcation in the π Contribution to the Electron Localization Function. ChemPhysChem 2008, 9, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.M.; Yoon, M.-C.; Lim, J.M.; Rath, H.; Naoda, K.; Osuka, A.; Kim, D. Reversal of Hückel (anti)aromaticity in the Lowest Triplet States of Hexaphyrins and Spectroscopic Evidence for Baird’s Rule. Nat. Chem. 2015, 7, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.M.; Oh, J.; Kim, W.; Mori, H.; Osuka, A.; Kim, D. Switching between Aromatic and Antiaromatic 1,3-Phenylene-Strapped[26]- and [28]Hexaphyrins upon Passage to the Singlet Excited State. J. Am. Chem. Soc. 2015, 137, 11856–11859. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.M.; Oh, J.; Cha, W.-Y.; Kim, W.; Lim, J.M.; Yoon, M.-C.; Kim, D. Control and Switching of Aromaticity in Various All-Aza-Expanded Porphyrins: Spectroscopic and Theoretical Analyses. Chem. Rev. 2017, 117, 2257–2312. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.; Krogh, E. Evidence for the Generationo of Aromatic Cationic Systems in the Excited State. Photochemical Solvolysis of Fluoren-9-ol. J. Chem. Soc. Chem. Commun. 1985, 17, 1207–1208. [Google Scholar] [CrossRef]
- Wan, P.; Krogh, E.; Chak, B. Enhanced Formation of 8π(4n) Conjugated Carbanions in the Excited State: First example of Photochemical C–H Bond Heterolysis in Photoexcited State. J. Am. Chem. Soc. 1988, 110, 4073–4074. [Google Scholar] [CrossRef]
- Wan, P.; Budac, D.; Krogh, E. Excited State Carbon Acids: Base Catalysed Photoketonization of Dibenzosuberenol to Dibenzosuberone via Initial C–H Bond Heterolysis from S1. J. Chem. Soc. Chem. Commun. 1990, 3, 255–257. [Google Scholar] [CrossRef]
- Wan, P.; Budac, D.; Earle, M.; Shukla, D. Excited-State Carbon Acid: Photochemical Carbon–Hydrogen Bond Heterolysis vs. Formal di-π-Methane Rearrangement of 5H-Dibenzo[a,c]cycloheptene and Related Compounds. J. Am. Chem. Soc. 1990, 112, 8048–8054. [Google Scholar] [CrossRef]
- Budac, D.; Wan, P. Excited-State Carbon Acid. Facile Benzylic Carbon–Hydrogen Bond Heterolysis of Subrene on Photolysis in Aqueous Solution: A Photogenerated Cyclically Conjugated 8π-electron Carbanion. J. Org. Chem. 1992, 57, 887–894. [Google Scholar] [CrossRef]
- Wan, P.; Shukla, D. Utility of Acid-Base Behavior of Excited States of Organic Molecules. Chem. Rev. 1993, 93, 571–584. [Google Scholar] [CrossRef]
- Mohamed, R.K.; Mondal, S.; Jorner, K.; Delgado, T.F.; Lobodin, V.V.; Ottosson, H.; Alabugin, I.V. The Missing C1–C5 Cycloaromatization Reaction: Triplet State Antiaromaticity Relief and Self-Terminating Photorelease of Formaldehyde for Synthesis of Fulvenes from Enynes. J. Am. Chem. Soc. 2015, 137, 15441–15450. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, R.; Li, H.; Bergman, J.; Lundstedt, A.; Jorner, K.; Ayub, R.; Haldar, S.; Jahn, B.O.; Denisova, A.; Zietz, B.; et al. Metal-free Photochemical Silylations and Transfer Hydrogenations of Benzenoid Hydrocarbons and Graphene. Nat. Commun. 2016, 7, 12962. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Jorner, K.; Sung, Y.M.; Mori, T.; Xiao, Q.; Kim, D.; Ottosson, H.; Aida, T.; Itoh, Y. Energetics of Baird Aromaticity Supported by Inversio of Photoexcited Chiral [4n]Annulene Derivatives. Nat. Commun. 2017, 8, 346. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Schleyer, P.R. Evaluation of Triplet Aromaticity by the Isomerization Stabilization Energy. Org. Lett. 2013, 15, 2442–2445. [Google Scholar] [CrossRef] [PubMed]
- An, K.; Zhu, J. Evaluation of Triplet Aromaticity by the Indene-Isoindene Isomerization Stabilization Energy Method. Eur. J. Org. Chem. 2014, 13, 2764–2769. [Google Scholar] [CrossRef]
- Ayub, R.; Papadakis, R.; Jorner, K.; Zietz, B.; Ottosson, H. The Cyclopropyl Group: An Excited State Aromaticity Indicator? Chem. Eur. J. 2017, 23, 13684–13695. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Naka, A.; Kobayashi, H. The Chemistry of Silacyclobutene: Synthesis, Reactions, and Theoretical Study. Coord. Chem. Rev. 2017, 335, 58–75. [Google Scholar] [CrossRef]
- Tzeng, D.; Fong, R.H.; Dilanjan, H.S.; Weber, W.P. Evidence for the Intermediacy of 1,1-dimethyl-2-phyenyl-1-sila-1,3-butadiene in the Photochemistry and Pyrolysis of 1,1-dimethyl-2-phenyl-1-sila-2-cyclobutene. J. Organomet. Chem. 1981, 219, 153–161. [Google Scholar] [CrossRef]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry. Part A: Structure and Mechanisms, 5th ed.; Springer: New York, NY, USA, 2007. [Google Scholar]
- Mauksch, M.; Tsogoeva, S.B. A Preferred Disrotatory 4n Electron Möbius Aromatic Transition State for a Thermal Electrocyclic Reaction. Angew. Chem. Int. Ed. 2009, 48, 2959–2963. [Google Scholar] [CrossRef] [PubMed]
- Brink, M.; Möllerstedt, H.; Ottosson, C.-H. Characteristics of the Electronic Structure of Diabatically and Adiabatically Z/E-Isomerizing Olefins in the T1 state. J. Phys. Chem. A 2001, 105, 4071–4083. [Google Scholar] [CrossRef]
- Villaume, S.; Ottosson, H. Aromaticity Changes along the Lowest-Triplet State Path for C=C Bond Rotation of Annulenyl-Substituted Olefins Probed by Electron Localization Function. J. Phys. Chem. A. 2009, 113, 12304–12310. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Fogarty, H.A.; Möllerstedt, H.; Brink, M.; Ottosson, H. Aromaticity Effects on the Profiles of the Lowest Triplet-State Potential-Energy Surfaces for Rotation about the C=C Bonds of Olefins with Five-Membered Ring Substituents: An Example of the Impact of Baird’s Rule. Chem. Eur. J. 2013, 19, 10698–10707. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Brink, M.; Möllerstedt, H.; Piqueras, M.C.; Crespo, R.; Ottosson, H. Z/E-Photoisomerizations of Olefins with 4nπ-or (4n + 2)π-Electron Substituents: Zigzag Variations in Olefin Properties along the T1 state Potential Energy Surfaces. J. Org. Chem. 2005, 70, 9495–9504. [Google Scholar] [CrossRef] [PubMed]
- Ottosson, H.; Eklöf, A.M. Silenes: Connectors between Classical Alkenes and Nonclassical Heavy Alkenes. Coord. Chem. Rev. 2008, 252, 1287–1314. [Google Scholar] [CrossRef]
- Kang, K.T.; Yoon, U.C.; Seo, H.C.; Kim, K.N.; Song, H.Y.; Lee, J.C. Thermal and Photochemical Reactions of Benzosilacyclobutenes with Alcohols. Intermediaxy of o-Silaquinone Methide in the Photochemical Reactions. Bull. Korean Chem. Soc. 1991, 12, 57–60. [Google Scholar]
- Bendikov, M.; Quadt, S.R.; Rabin, O.; Apeloig, Y. Addition of Nucleophiles to Silenes. A Theoretical Study of the Effect of Substituents on Their Kinetic Stability. Organometalllics 2002, 21, 3930–3939. [Google Scholar] [CrossRef]
- Kang, K.T.; Seo, H.C.; Kim, K.N. Thermal Reactions of Benzosilacyclobutenes with Alcohols. Tetrahedron Lett. 1985, 26, 4761–4762. [Google Scholar] [CrossRef]
- Walsh, R. Bond Dissociation Energy Values in Silicon-Containing Compounds and Some of their Implications. Acc. Chem. Res. 1981, 14, 246–252. [Google Scholar] [CrossRef]
- McMillen, D.F.; Golden, D.M. Hydrocarbons Bond Dissociation Energies. Ann. Rev. Phys. Chem. 1982, 33, 493–532. [Google Scholar] [CrossRef]
- Segura, J.L.; Martín, N. o-Quinodimethanes: Efficient Intermediates in Organic Synthesis. Chem. Rev. 1999, 99, 3199–3246. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Naka, A.; Yoshizawa, K. The Chemistry of Benzodisilacyclobutenes and Benzobis(disilacyclobutene)s: New Development of Transition-Metal-Catalyzed Reactions, Stereochemistry and Theoretical Studies. Dalton Trans. 2016, 45, 3210–3225. [Google Scholar] [CrossRef] [PubMed]
- Taubert, S.; Sundholm, D.; Jusélius, J. Calculations of Spin-Current Densities Using Gauge-Including Atomic Orbitals. J. Chem. Phys. 2011, 134, 54123–54135. [Google Scholar] [CrossRef] [PubMed]
- Soncini, A.; Fowler, P.W. Ring-Current Aromaticity in Open-Shell Systems. Chem. Phys. Lett. 2008, 450, 431–436. [Google Scholar] [CrossRef]
- Jursic, B.S. Exploring the lowest energy triplet potential energy surface for cyclic c4h4 isomers with the complete basis set ab initio method. Is the transformation of triafulvene into cyclobutadiene possible in their excited states? J. Mol. Struct. (THEOCHEM) 1999, 490, 133–144. [Google Scholar] [CrossRef]
- Brook, M.A. Silicon in Organic, Organometallic, and Polymer Chemistry; John Wiley and Sons: Hoboken, NJ, USA, 2000; ISBN 0-471-19658-4. [Google Scholar]
- Zamstein, N.; Kallush, S.; Segev, B. A phase-space approach to the T1 ⇝ S0 radiationless decay in benzene: The effect of deuteration. J. Chem. Phys. 2005, 123, 074304. [Google Scholar] [CrossRef] [PubMed]
- Schaffroth, M.; Gershoni-Poranne, R.; Stanger, A.; Bunz, U.H.F. Tetraazaacenes Containing Four-Membered Rings in Different Oxidation States. Are They Aromatic? A Computational Study. J. Org. Chem. 2014, 79, 11644–11650. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.R.; Pühlhofer, F. Recommendations for the Evaluation of Aromatic Stabilization Energies. Org. Lett. 2002, 4, 2873–2876. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R.; Chang, H.W.; Hill, R.; Wasserman, E. Stable Triplet States of Some Cyclopentadienyl Cations. J. Am. Chem. Soc. 1967, 89, 1112–1119. [Google Scholar] [CrossRef]
- Saunders, M.; Berger, R.; Jaffe, A.; McBride, J.M.; O’Neill, J.; Breslow, R.; Hoffman, J.M., Jr.; Perchonock, C.; Wasserman, E.; Hutton, R.S.; et al. Unsubstituted Cyclopentadienyl Cation, a Ground-State Triplet. J. Am. Chem. Soc. 1973, 95, 3017–3018. [Google Scholar] [CrossRef]
- Wörner, H.J.; Merkt, F. Photoelectron Spectroscopic Study of the First Singlet and Triplet States of the Cyclopentadienyl Cation. Angew. Chem. Int. Ed. 2006, 45, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Wörner, H.J.; Merkt, F. Diradicals, Antiaromaticity, and the Pseudo-Jahn–Teller Effect: Electronic and Rovibronic Structures of the Cyclopentadienyl Cation. J. Chem. Phys. 2007, 127, 34303. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculations of Vibrational Absorption and Circular Dichorism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. A New Mixing of Hartree–Fock and Local-Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Handy, N.C.; Cohen, A.J. Left-Right Correlation Energy. Mol. Phys. 2001, 99, 403–412. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Rrishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Schleyer, P.R. Introduction: Aromatcitiy. Chem. Rev. 2001, 101, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Krygowski, T.M. Crystallographic Studies of Inter- and Intramolecular Interactions Reflected in Aromatic Character of π-Electron Systems. J. Chem. Inf. Comput. Sci. 1993, 33, 70–78. [Google Scholar] [CrossRef]
- Stanger, A. Nucleus-Independent Chemical Shifts (NICS): Distance Dependence and Revised Criteria for Aromaticity and Antiaromaticity. J. Org. Chem. 2006, 71, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Stanger, A. Obtaining Relative Induced Ring Currents Quantitatively from NICS. J. Org. Chem. 2010, 75, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- Rahalkar, A.; Stanger, A. Aroma. Available online: http://schulich.technion.ac.il/Amnon_Stanger.htm (accessed on 22 November 2015).
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Herges, R.; Geuenich, D. Delocalization of Electrons in Molecules. J. Phys. Chem. A 2001, 105, 3214–3220. [Google Scholar] [CrossRef]
- Geuenich, D.; Hess, K.; Kohler, F.; Herges, R. Anisotropy of the Induced Current Density (ACID), a General Method to Quantify and Visualize Electronic Delocalization. Chem. Rev. 2005, 105, 3758–3772. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A.; Bader, R.F.W. Calculations of Magnetic Response Properties Using a Continuous Set of Gauge Transformations. Chem. Phys. Lett. 1993, 210, 223–231. [Google Scholar] [CrossRef]
- Yan, D.; Mohsseni-Ala, J.; Auner, N.; Bolte, M.; Bats, J.W. Molecular Optical Switches: Synthesis, Structure, and Photoluminescence of Spirosila Compounds. Chem. Eur. J. 2007, 13, 7204–7214. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | S0 | T1 | ||||
|---|---|---|---|---|---|---|
| a | b | ∆HOMA | a | b | ∆HOMA | |
| 1 | −4.04 | −1.11 | 2.93 | 0.10 | −0.59 | −0.69 |
| 2 | 0.97 | 0.07 | −0.90 | −0.32 | 0.83 | 1.15 |
| 3 | 0.08 | −0.21 | −0.13 | 0.89 | 0.21 | −0.68 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayub, R.; Jorner, K.; Ottosson, H. The Silacyclobutene Ring: An Indicator of Triplet State Baird-Aromaticity. Inorganics 2017, 5, 91. https://doi.org/10.3390/inorganics5040091
Ayub R, Jorner K, Ottosson H. The Silacyclobutene Ring: An Indicator of Triplet State Baird-Aromaticity. Inorganics. 2017; 5(4):91. https://doi.org/10.3390/inorganics5040091
Chicago/Turabian StyleAyub, Rabia, Kjell Jorner, and Henrik Ottosson. 2017. "The Silacyclobutene Ring: An Indicator of Triplet State Baird-Aromaticity" Inorganics 5, no. 4: 91. https://doi.org/10.3390/inorganics5040091
APA StyleAyub, R., Jorner, K., & Ottosson, H. (2017). The Silacyclobutene Ring: An Indicator of Triplet State Baird-Aromaticity. Inorganics, 5(4), 91. https://doi.org/10.3390/inorganics5040091